Neuromusculaire en neurocutane ziektenadmin22 april, 2010 - 10:27
Myotone dystrofie type 1
Myotone dystrofie type 1fajenn25 juni, 2010 - 16:41
Inleiding
Inleiding
Myotone dystrofie type 1 (MD1) is een complexe, zeer langzaam progressieve, erfelijke ziekte. “Myotoon” wijst op vertraagd ontspannen van skeletspieren na aanspanning en ”dystrofie” op onvoldoende volume van skeletspieren. De toevoeging “type 1” geeft aan dat er een aan MD1 verwante ziekte is (MD2). (De Die-Smulders et al. 2010; Jennekens-Schinkel & Jennekens 2008). Hoewel de naam van de ziekte dus past op een spieraandoening, staan bij kinderen jonger dan 10 jaar stoornissen van cognitie en gedrag mede op de voorgrond.
admin30 maart, 2009 - 18:36
Andere teksten op internet
Andere teksten op internet
www.erfelijkheid.nl. Deze site biedt afzonderlijk informatie over MD1 voor (para)medici en voor patiënten. De site wordt gesteund door de Nederlandse Centra voor Klinische Genetica. www.vsn.nl. Via de website van de Vereniging Spierziekten Nederland kan men informatie verkrijgen over myotone dystrofie type 1, ook over de aangeboren en kindervormen van de ziekte. www.kinderneurologie.eu geeft een uitvoerige beschrijving van de ziekte en over de behandeling van ziekteverschijnselen. De tekst is bestemd voor hulpverleners en familieleden. www.neurologie.nl biedt toegang tot een richtlijn over myotone dystrofie bij volwassenen (18 jaar en ouder).
fajenn25 juni, 2010 - 16:44
Epidemiologie
Epidemiologie
Onderzoek over de prevalentie in ons land ontbreekt. Mede op grond van gegevens uit andere landen wordt de prevalentie van MD1 in Nederland geschat op 5-15 per 100.000 mensen (de Die-Smulders et al., 2010; www.vsn.nl). Uitgaande van deze cijfers zouden er in Nederland ongeveer 1000-2000 mensen met MD1 zijn. Het aantal kinderen met de ziekte bedraagt waarschijnlijk enige honderden. Of de ziekte bij allochtonen evenveel voorkomt als bij autochtonen is onbekend.
fajenn25 juni, 2010 - 16:45
Oorzaak
Oorzaak
Genmutatie
In een gen op de lange arm van chromosoom 19 ((19q13.3) bevindt zich, in een deel dat voor de coderende functie van het gen niet van belang is, een aantal kopieën van een drietal (een triplet) verschillende nucleotiden die de organische basen cytosine, thymine en guaninine bevatten. Men spreekt daarom van een CTG triplet. Het aantal kopieën (CTG-tripletten) varieert normaal van 5 tot 37 en bij mensen met MD1 van ongeveer 50 tot meer dan 3000 (de Die-Smulders et al., 2010). Hoe groter het aantal kopieën, hoe eerder het debuut van de ziekte en hoe ernstiger de verschijnselen. In opeenvolgende generaties van mensen met de ziekte neemt het aantal kopieën toe.
Het afwijkende gen vertaalt zich in afwijkingen van het RNA. Het gemuteerde RNA hoopt zich op in de kernen en heeft daar een storend effect.
Hersenafwijkingen
Er zijn verschillende - onder de microscoop zichtbare - veranderingen gerapporteerd waarvan niet bekend is of ze algemeen voorkomen en waarvan de betekenis voor de neurologische en neuropsychologische stoornissen onduidelijk is. Met een MRI techniek (DTI, diffusion tensor imaging, zie beeldvorming) zijn consistente afwijkingen in de witte stof zichtbaar gemaakt, zowel bij de kindervormen van de ziekte als bij volwassenen. Een relatie tussen deze witte stof afwijkingen en de cognitieve stoornissen is aannemelijk gemaakt Wozniak et al., 2011; Minnerop et al., 2011)
fajenn27 mei, 2011 - 13:28
Erfelijkheid
Erfelijkheid
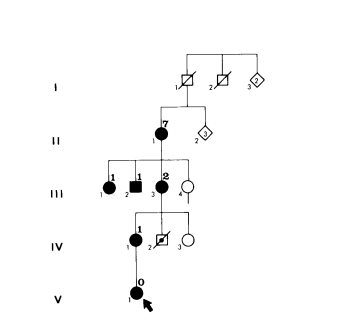
Ieder kind van een zieke ouder heeft een kans van 50% op de aandoening. Men noemt deze vorm van overdracht van een ziekte “autosomaal dominante overerving”. Bij MD 1 bestaat anticipatie, (zie figuur stamboom): In opeenvolgende generaties van mensen met MD1 treden de verschijnselen eerder in het leven op. In de eerste generatie zijn de verschijnselen gering en ze manifesteren zich pas op latere leeftijd. In latere generaties zijn de verschijnselen ernstiger en blijken ze vroeger in het leven. Tenslotte zijn de ziekteverschijnselen er vanaf de geboorte. Kinderen met de congenitale, ernstigste vorm van de ziekte worden meestal geboren uit moeders met de ziekte (“maternale” overerving). Een abnormale herhaling (“repeat”) van drie bouwelementen (de nucleotiden cytosine, thymine en guanine) in een gen op chromosoom 19 (19q. 13.3) ligt aan de ziekte ten grondslag. Hoe groter het aantal herhalingen, des te eerder het begin (debuut) van de ziekteverschijnselen. Figuur 1. Stamboom van Familie C. Overgenomen uit de dissertatie van CJ Höweler, 1986.
I.De man I1 was overleden op het tijdstip dat deze familie werd onderzocht. Hij zou geen verschijnselen van MD1 hebben gehad. Zijn broer I2 zou wel verschijnselen zoals voorkomend bij MD1 hebben gehad.
II.De vrouw II1 was bij onderzoek in het 7de decennium (tussen 60 en 70 jaar oud); zij had cataract (staar) en verschijnselen van myotonie.
III.De vrouw III3 had duidelijke verschijnselen van de klassieke vorm van MD1. De vrouw III1 en de man III.2 hadden myotonie sedert de kinderjaren en inmiddels ook spierzwakte; ze waren opvallend passief.
IV.De vrouw IV1 had de kindervorm van MD1, ze was zwakzinnig en sprak dysarthrisch. De jongen IV2 was overleden, waarschijnlijk ten gevolge van de congenitale vorm van MD1.
V.Het meisje V1 was overleden ten gevolge van ernstige verschijnselen van de congenitale vorm van MD1.
Verklaring: Romeinse cijfers = generaties; grote Arabische cijfers boven gesloten symbolen = levensdecennium waarin MD1 zich openbaarde (leeftijd tussen 60 en 70 jaar); kleine cijfers onder symbolen = persoonsnummer in de studie; vierkant = man; rondje = vrouw; ruit met inwendig cijfer = aantal niet onderzochte gezinsleden; zwarte vulling = MD1 gediagnosticeerd; diagonale streep = overleden; stip in symbool met diagonale streep = dood bij geboorte
admin16 april, 2009 - 14:22
Ziekteverschijnselen
Ziekteverschijnselen
Tabel.De belangrijkste verschijnselen van de vier vormen van MD1
intelligentie laag normaal of lichte tot matige zwakzinnigheid, zwakte van gelaat- en keelspieren, onduidelijke spraak, gedragstoornis, vanaf 5de - 10de jaar geleidelijk verschijnselen zoals bij klassieke vorm
Vanaf de geboorte – congenitale vorm
spierslapte bij geboorte, zwakte van gelaat- en keelspieren, ernstige zwakzinnigheid tot laag normale intelligentie, gedragstoornis, vanaf 5de tot 10de jaar geleidelijk verschijnselen zoals bij klassieke vorm
Zwakte van de gelaatspieren
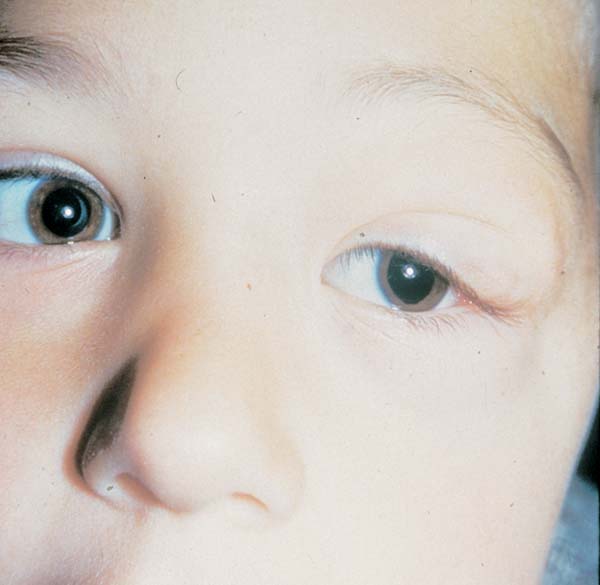
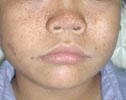
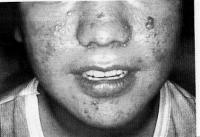
Zwakte van gelaat- en keelspieren is vooral bij kinderen met de congenitale vorm maar ook bij kinderen met debuut op kinderleeftijd vroegtijdig aanwezig. Als gevolg daarvan ontwikkelt de aangezichtschedel abnormaal (zie Figuur 2). Het onderste deel van het gelaat is te lang en het gehemelte is hoog en smal. Het gelaat is slap, de mimiek is onvoldoende en de spraak is moeilijk verstaanbaar. Zes en vijftig (56) kinderen en adolescenten (36 met de congenitale vorm, 18 met de kindervorm en twee met de klassieke vorm) hadden allen onvoldoende gelaatsexpressie; bij de meeste kinderen hing de mond in rust open en 28 kinderen hadden dysarthrie (moeilijk verstaanbare spraak) (Sjögreen et al. 2007). Kinderen met MD1 hebben meer plaque, meer tandvleesontsteking en meer cariës dan leeftijdsgenoten (Engvall et al., 2007).
Figuur 2. Zwakte van gelaatspieren bij congenitale vorm van myotone dystrofie type 1.
Figuur overgenomen van Jennekens et al., 2000, p 18
[img_assist|nid=922|title=Figuur 2. Zwakte van gelaatspieren bij congenitale vorm van myotone dystrofie type 1.|desc=|link=popup|align=center|width=466|height=640]Figuur overgenomen van Jennekens et al., 2000, p 18
Slaapzucht
Kinderen hebben wat meer slaap nodig dan gebruikelijk, maar “slaapzucht” is, anders dan bij volwassenen met MD1, niet zeer opvallend (Quera Salva et al. 2006).
De diagnose
De diagnose kan definitief worden gesteld door het aantonen van de genetische afwijking. Dit onderzoek wordt verricht als de ziekte- verschijnselen en gegevens over het voorkomen van de ziekte bij een van de ouders daar aanleiding toe geven.
Beloop en medische behandeling
De moeilijke start van kinderen met de congenitale vorm van de ziekte is in variabele mate het gevolg van ademhalingszwakte, slecht zuigen en slikken, onvoldoende motoriek van het maag-darmstelsel en contracturen. Deze verschijnselen kunnen ieder in zekere mate worden opgevangen of tegengegaan door ademhalingsondersteuning, maagsonde voeding, intraveneuze voeding en passief buigen en strekken van gewrichten. De verschijnselen bij de kindervorm van de ziekte kunnen zich gedurende enige tijd beperken tot verminderde mimiek, onvoldoende verstaanbaarheid van de spraak en - vaak - leerstoornissen. Heel geleidelijk ontwikkelen zich zowel bij de congenitale als de kindervorm de verschijnselen van de klassieke vorm. De levensduur wijkt bij de kindervorm en congenitale vorm waarschijnlijk niet veel af van die bij de klassieke vorm, maar hierover zijn nog onvoldoende gegevens. Behandeling van de oorzaak van de ziekte is nog niet mogelijk, wel die van veel ziekteverschijnselen. Denk bijvoorbeeld aan correctie van scheelzien, tandheelkundige zorg, aanpassing van het dieet bij eventuele slikproblemen en stimulering van de stoelgang. Myotone dystrofie type 1 is berucht vanwege de kans op ernstige complicaties bij algemene narcose.
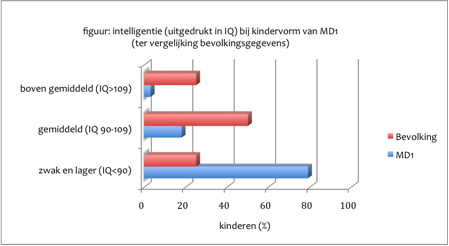
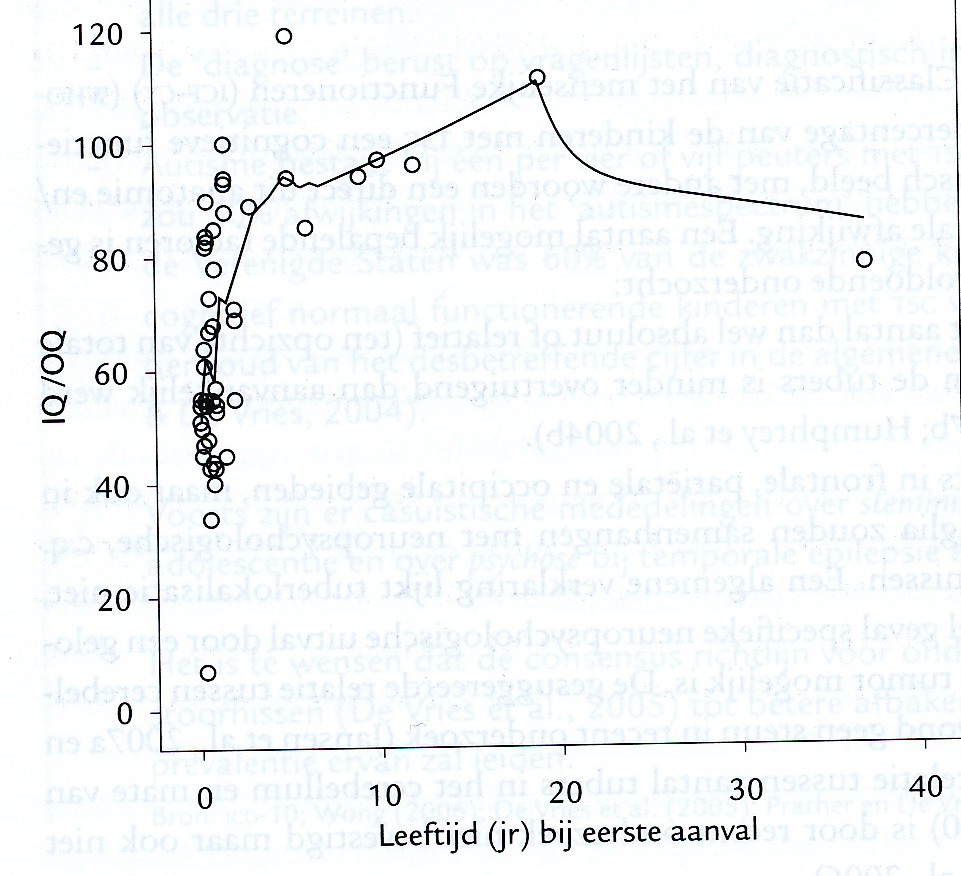
Het Totale intelligentiequotiënt (IQ) varieert van lager dan 35 (ernstige zwakzinnigheid) tot – bij uitzondering – boven gemiddeld. Ernstige zwakzinnigheid (grenswaarde van het IQ voor matige en ernstige zwakzinnigheid 50) komt voor bij de congenitale vorm van MD1, vooral bij kinderen met een na de geboorte erg moeilijke start (ENCYCL-Zwakzinnigheid). Slechts weinig kinderen met de congenitale vorm hebben een laag normale intelligentie (IQ varieërt van meer dan 70 tot 85) en heel weinige een normale intelligentie (Ekström et al. 2008; 2009). Bij de kindervorm van MD1 is het Totale IQ gemiddeld lager dan in de gehele bevolking. Een gemiddelde heeft voor het individuele kind weinig betekenis wanneer de spreiding zo groot is als bij deze vorm van MD1 (zie figuur, ontleend aan Angeard et al. 2007). Zo was in een groep van 34 kinderen het Totale IQ gemiddeld 69,8. Maar het laagste IQ was 42, passend bij matige zwakzinnigheid. En het hoogste 114, passend bij een boven gemiddelde intelligentie (Angeard et al. 2007). Eerder was bij 24 kinderen een niet wezenlijk ander beeld gevonden (Steyaert et al. 2000).
Staafdiagram op basis van gegevens van Angeard et al., 2007
[img_assist|nid=1208|title=Staafdiagram op basis van gegevens van Angeard et al., 2007 |desc=|link=popup|align=center|width=480|height=263] Het Verbale IQ is bij de kindervorm misschien iets hoger dan het Performale IQ maar het verschil is niet groot en het aantal kinderen waarvoor deze gegevens beschikbaar zijn is beperkt zodat een definitieve uitspraak nog niet mogelijk is. Bij overerving via de moeder is het Totale IQ gemiddeld wat lager dan bij paternale (via de vader) overerving.
Specifieke tekorten in andere domeinen van het cognitief functioneren zijn niet gevonden of nog onvoldoende bekend om conclusies te kunnen trekken. Geheugen en leervermogen zijn naar verhouding sterk. Traagheid van denken is geopperd maar niet bevestigd (zie Jennekens-Schinkel en Jennekens, 2008).
Tot de kernverschijnselen van autisme behoren onvoldoende sociale interactie, onvoldoende communicatie en stereotyp (telkens terugkerend) gedrag (ENCYCL-Autisme). Voor het vaststellen van autisme bestaat geen laboratoriumproef die de resultaten van vragenlijstonderzoek, interview en algemene indruk kan toetsen (Spence & Thurm, 2010). Dat vergroot de kans op een subjectief element bij het vaststellen van de stoornis. Kinderen met congenitale MD1 communiceren moeilijk door de gelaatspierzwakte en de spraakstoornis (dysartrie). In één studie werd tot autisme geconcludeerd bij iets meer dan één op de twee kinderen met de congenitale vorm van MD1 en bij één op de zes kinderen met de kindervorm, vooral op grond van onvoldoende sociale interactie en onvoldoende communicatie (Ekström et al. 2008). Het zou kunnen dat de criteria van autisme niet scherp zijn onderscheiden van de verschijnselen van MD1.
Stoornis in temperament
Volwassenen met MD1 hebben volgens hun omgeving weinig initiatief, zij zijn weinig fel in hun reageren op prikkels en zij hebben een laag energie- en activiteitsniveau. Zij hebben, kortom, een nog niet begrepen afwijking van het temperament (het hoe van gedrag, de gedragsstijl). Op een temperamentlijst scoorden zij extreem laag wanneer het ging om behoefte aan nieuwe ervaringen en avontuur. Andere dimensies van het temperament (extraversie, emotionaliteit, impulsiviteit) waren ongestoord. (Jennekens-Schinkel & Jennekens, 2008).
Het gedrag van kinderen wordt beschreven als kalm, neigend tot passiviteit, ze nemen weinig initiatieven voor het komen tot contact, ze zijn niet snel afgeleid, ze passen zich goed aan en ze zijn vriendelijk (Ekström et al., 2010; Steyaert et al., 1997)
admin9 november, 2009 - 15:03
Psychosociaal functioneren
Psychosociaal functioneren
Schoolloopbaan
Omstreeks 50% of minder van de kinderen met de kindervorm van MD1 kan met extra onderwijskundige hulp regulier onderwijs volgen. De overigen behoeven speciaal onderwijs evenals kinderen met de congenitale vorm (Quera Salva et al., 2006).
Sociale interactie
De slapte van het gezicht doet afbreuk aan de mogelijkheden tot het uiten van emoties. De onduidelijkheid van de spraak belemmert de communicatie. In sport scoren de kinderen slecht. Hun passiviteit en - bij sommigen - de verhoogde slaapbehoefte worden niet altijd begrepen als ziekteverschijnselen. Autisme kan het tekort in interactie nog versterken. Meer dan andere kinderen staan zij bloot aan pesterijen.
admin9 november, 2009 - 15:17
Begeleiding
Begeleiding
De combinatie van lichamelijke-, cognitieve- en gedragstoornissen, en het uiterst langzame progressieve beloop stellen hoge eisen aan de begeleiding.
Ouders kunnen zich bezwaard voelen door het besef van hun verantwoordelijkheid voor de ziekte van hun kind en kunnen om die reden bijstand nodig hebben. Wat ook de beperkingen van de kinderen zijn, ze moeten zoveel als mogelijk tot zelfstandige mensen worden opgevoed
Ouders, leraren en andere begeleiders behoeven informatie over de vele ongewone aspecten van de ziekte: de gevolgen van de spierzwakte in het gelaat voor mimiek en verstaanbaarheid van de spraak, de kans op zeer verschillende lichamelijke klachten, de passieve instelling van de kinderen, de meerdere slaapbehoefte van sommige kinderen, de beperkingen die de ziekte geeft in sport en spel, het risico dat de kinderen worden gepest en het risico dat de kinderen ten onrechte als lui worden beschouwd.
Onderzoek van de cognitie kan gewenst zijn voor een verantwoord advies over onderwijs en schoolkeuze. De gelaatsuitdrukking van kinderen kan een onjuiste schatting van de cognitieve mogelijkheden in de hand werken.
Begeleiders moeten zo nodig ouders stimuleren het kind tenminste eenmaal per jaar medisch te laten evalueren.
admin9 november, 2009 - 15:18
Literatuur
Literatuur
Angeard N, Gargiulo M, Jacquette A, Radvanyi H, Eymard B, Héron D. (2007) Cognitive profile in childhood myotonic dystrophy type 1: Is there a global impairment? Neuromuscular Disorders 17: 451-458
De Die-Smulders CEM, Jennekens FGI, Faber CG (2010) Myotonic dystrophy type 1. In: Cassidy SB, Allanson JE (redacteuren) Management of genetic syndromes. 3de druk, Wiley & Sons Inc. pp 529-547
Ekström AB, Hakenäs-Plate L, Samuelsson L, Tulinius M, Wentz E. (2008) Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. American Journal of Medical Genetics B Neuropsychiatric Genetics 147B : 918-926
Ekström AB, Hakenās-Plate L, Tulinius M, Wentz E (2009) Cognition and adaptive skills in myotonic dystrophy type 1: a study of 55 individuals with congenital and childhood forms. Developmental Medicine & Child Neurology 51: 982-990
Engvall M, Sjögreen L, Kjellberg H, Robertson A, Sundall S, Killarides S. (2007). Oral health in children and adolescents with myotonic dystrophy. European Journal of Oral Science 15; 192-197
Jennekens FGI, De Die-Smulders CEM, Busch HFM, Höweler CJ. (2000) Myotone dystrofie begeleiding en behandeling. Maarssen: Elsevier Gezondheidszorg
Jennekens-Schinkel A, Jennekens FGI. (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam: Uitgeverij Boom, pp 575-590
Minnerop M en 10 andere auteurs. (2011) The brain in myotonic dystrophy type 1 and 2: evidence for a predominant white matter disease. Brain 134: 3530-3546
Quera Salva M-A, Blumen M, Jacquette A, Durand M-C, Andre S, Villiers M de et al. (2006) Sleep disorders in childhood-onset myotonic dystrophy type 1. Neuromuscular Disorders 2006; 16: 564-570
Sjögreen L, Engvall M, Ekström A, Lohmander A, Kiliaridis S, Tulinius M. (2007) Orofacial dysfunction in children and adolescence with myotonic dystrophy. Developmental Medicine and Child Neurology 2007; 49: 18-22
Spence SJ, Thurm A. (2010) Testing autism interventions:trials and tribulations. Lancet 375: 2124-2125
Steyaert J, De Die-Smulders C, Frijns J-P, Goossens E, Willekens D. (2000) Behavioral phenotype in childhood type of dystrophia myotonica. American Journal of Medical Genetics (Neuropsychiatric Genetics) 96: 888-889
Wozniak JR, Mueller BA, Ward EE, Lim KO, Day JW. (2011) White matter abnormalities and neurocognitive correlates in children and adolescents with myotonic dystrophy type 1: a diffusion tensor imaging study. Neuromuscular Disorders 21: 89-96
Duchenne spierdystrofie ("Duchenne muscular dystrophy", afgekort DMD) is een langzaam verergerende erfelijke ziekte die vrijwel uitsluitend voorkomt bij jongens en mannen. De aandoening beperkt zich niet tot de skeletspieren, ook in hartspier en hersenen ontstaan afwijkingen. Afhankelijk van de behandeling kan de ziekteduur tientallen jaren bedragen.
Na korte beschrijvingen van epidemiologie, erfelijkheid, ontstaanswijze, lichamelijke verschijnselen en medische behandeling, geeft de tekst informatie over cognitie, psychosociale factoren en begeleiding.
fajenn26 januari, 2011 - 22:52
Andere teksten op internet
Andere teksten op internet
www.erfelijkheid.nl biedt een goed, bondig overzicht. www.vsn.nl biedt veel en verschillende informatie, ook over sociale aangelegenheden.
Via de website van de Vereniging Spierziekten Nederland (VSN) kan men een richtlijn voor hulpverleners over behandeling en begeleiding oproepen. De richtlijn dateert van 2002. www.dmd.nl biedt een uitvoerig Engelstalig overzicht van erfelijke aspecten en lichamelijke verschijnselen.
fajenn26 januari, 2011 - 22:53
Epidemiologie
Epidemiologie
Doorgaans wordt vermeld dat de ziekte in Nederland ontstaat bij één per 4.000 jongens of dat vijf per 100.000 mannelijke personen er aan lijden. Deze cijfers dateren van begin jaren negentig in de vorige eeuw (Van Essen et al., 1992). Dankzij erfelijkheidsadvisering zijn de huidige cijfers waarschijnlijk wat gunstiger. Zo komt de ziekte tegenwoordig nog maar zelden bij meer dan één jongen in een gezin voor.
fajenn26 januari, 2011 - 22:55
Erfelijkheid en genmutatie
Erfelijkheid en genmutatie
Duchenne spierdystrofie is een geslachtsgebonden aandoening; de ziekte is het gevolg van een mutatie in één enkel gen, het gen dat de code bevat voor het eiwit dystrofine op de korte arm van het X-chromosoom. Meisjes met een dergelijke genmutatie hebben geen of weinig spierzwakte doordat zij twee X-chromosomen hebben; het normale dystrofine gen op het tweede X-chromosoom biedt meestal voldoende compensatie om ziekteverschijnselen te voorkomen. Jongens ontwikkelen wel spierzwakte doordat zij maar één X-chromosoom hebben. Bij hen ontbreekt dystrofine in spierweefsel.
Ieder kind van een draagster van een gemuteerd Duchenne gen heeft een kans van 50% om het afwijkende gen te erven.
Er zijn tal van verschillende mutaties in het dystrofine gen bekend. Sommige mutaties leiden tot ontbreken van dystrofine, zoals bij de ziekte van Duchenne, andere tot afwijkingen van het dystrofine. Dystrofine-afwijkingen hebben ook spierzwakte tot gevolg, maar die ontstaat later dan bij de ziekte van Duchenne en is minder ernstig. Men spreekt in dit geval van de ziekte van Becker.
fajenn27 januari, 2011 - 10:19
Het ontstaan
Het ontstaan
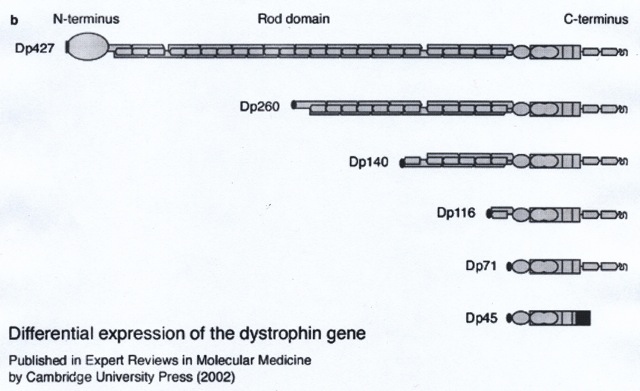
Dystrofine is een groot staafvormig proteïne (eiwit) met een gewicht van 427 kilodalton (afgekort Dp427). In dwarsgestreepte spiervezels bevindt dystrofine zich verspreid onder de spiervezelmembraan. Bij ontbreken ervan in spierweefsel bestaat risico op beschadiging van de spiervezelmembranen tijdens contractie. Het risico is het grootst voor lange vezels.
Dp427 is ook aangetoond in zenuwcellen van de hersenschors en de hippocampus, en in de Purkinjecellen van het cerebellum (ENCYCL-Anatomie van de hersenen). In hersencellen komen ook veel kortere vormen van dystrofine – Dp140 en vooral Dp71 – voor.Figuur 1. Schematische weergave van dystrofine isovormen.
Deze korte vormen ontstaan doordat alleen het C-terminale distale deel van het gen wordt afgelezen. Duchenne-mutaties in het proximale deel van het gen leiden tot ontbreken van het grote Dp427 en lijken van weinig betekenis voor de cognitie te zijn. Distale gen mutaties leiden tot functieverlies van alle vormen. Er zijn steeds meer aanwijzingen dat zwakzinnigheid in het bijzonder voorkomt bij deze distale mutaties (Ricotti et al., 2011; Taylor et al., 2010). Over de lokalisatie en functie van Dp140 en Dp71 in de hersenen is nog weinig bekend.
fajenn27 januari, 2011 - 10:14
Lichamelijke verschijnselen
Lichamelijke verschijnselen
Diagnostische fase
Spierzwakte manifesteert zich het eerst aan bovenbenen, bekken en rug.
Baby’s met Duchenne spierdystrofie gaan op tijd lachen.
Bijna 40% van de peuters met Duchenne spierdystrofie zit laat zonder steun en 70% is laat met ongesteund lopen (Cyrulnik et al., 2007).
Kleuters hebben moeite met gaan staan, kunnen niet hardlopen of hinkelen, lopen te veel op de tenen en vallen veel.
Diagnose
Bij bloedonderzoek worden extreem hoge waarden voor de activiteit van het spierenzym creatine fosfokinase gevonden. DNA-onderzoek toont de mutatie in het dystrofine-gen (locatie Xp21). De mutatie is niet altijd eenvoudig vindbaar doordat het gen groot is en de mutatie klein kan zijn (Bushby et al., 2010). Lukt het niet, of is er onzekerheid over het onderscheid tussen de Duchenne- of Becker- vormen van spierdystrofie, dan moet een spierbiopt worden genomen voor onderzoek naar dystrofine (De Die-Smulders et al., 2004; zie ook www.erfelijkheid.nl).
Ziektebeloop
Door abnormaal verschil in kracht tussen buig- en strekspieren ontstaan contracturen, bijvoorbeeld spitsvoetstand.
Tussen 7 en 13 jaar worden de jongens rolstoelafhankelijk. In de rolstoel ontstaat meestal ernstige verkromming (scoliose) van de wervelkolom. Om deze te voorkomen is vaak operatie nodig.
Adolescenten behoeven vanaf ongeveer het 16de levensjaar of later ademhalingsondersteuning.
Enige spierkracht blijft het langst aanwezig in de duimmuis en in de gelaatspieren. De oogbewegingen blijven behouden.
Voeding kan, als slikken onvoldoende of niet meer mogelijk is, plaatsvinden via een opening in de buikwand (percutane endoscopische gastrostomie , afgekort PEG of percutane radioscopische gastrostomie, afgekort PRG).
Ademhalingsondersteuning
Aanvankelijk wordt de ademhaling alleen ’s nachts ondersteund; het functioneren overdag is dan beter. Ademhalingsondersteuning overdag is (in eerste instantie) mogelijk via een mondstuk dat met de hand in de mond wordt gehouden. Wanneer de armfunctie daarvoor niet goed genoeg meer is, kan beademing geschieden via een daartoe aangelegde opening in de luchtpijp (tracheostomie). Bij beademing via een tracheostoma blijft spreken mogelijk en kan slijm eenvoudig uit de luchtpijp worden weggezogen (Dreyer et al., 2010; Meinesz et al., 2007).
Levensduur
Zonder ademhalingsondersteuning volgt overlijden doorgaans omstreeks het 19de jaar of eerder, met tracheostomale ademhalingsondersteuning tussen het 25ste en 40ste jaar of later. Tot de geregistreerde overlijdensoorzaken bij beademing behoren onder andere hartritmestoornissen en hartzwakte (Meinesz et al., 2007).
Behandeling met corticosteroïden en gevolgen voor het zelfbeeld
Behandeling met prednison kan verlies van spierkracht vertragen: rolstoelafhankelijkheid ontstaat later, scolioseoperaties zijn minder vaak nodig, ademhalingsondersteuning kan later beginnen en het effect op de hartspier is gunstig. Langdurige behandeling met prednison leidt tot veel bijwerkingen. Verschillende hiervan hebben gevolgen voor het (zelf)beeld van Duchenne jongeren, te weten verminderde groei, gewichtstoename, vollemaansgezicht, acne. Euforie of depressie kunnen tot gedragsveranderingen leiden. Een aanmerkelijke minderheid (in de Verenigde Staten 25%) van de jongens staakt na enige tijd de behandeling, meestal vanwege de gewichtstoename (Moxley et al., 2010 ).
fajenn27 januari, 2011 - 12:39
Cognitie en gedrag
Cognitie en gedrag
Intelligentiequotiënt
Uit een groot aantal onderzoekingen blijkt dat gemiddeld het Totale intelligentiequotiënt (TIQ) van jongens met Duchenne spierdystrofie tussen 80 en 85 ligt, ongeveer 1 standaarddeviatie lager dan in de algemene bevolking (Taylor et al., 2010; Cotton et al., 2001). Ongeveer 25 tot 35% van de jongens heeft een TIQ lager dan 70, de grenswaarde voor lichte zwakzinnigheid. Ter vergelijking: in de algemene bevolking behaalt 2% een TIQ lager dan 70. Totale IQs lager dan 50, passend bij matige of diepere zwakzinnigheid, zijn bij Duchenne spierdystrofie uitzonderingen. Bij 7–8% van de jongens met Duchenne spierdystrofie is het TIQ trouwens hooggemiddeld of bovengemiddeld (boven 110). Kortom, wat betreft intelligentie is Duchenne spierdystrofie een aandoening met grote heterogeniteit.
Verbaal IQ en Performaal IQ
Het Verbale IQ is veelal wat (gemiddeld 5–7 punten) lager dan het Performale IQ. Verschillen tussen de beide componenten van het IQ kunnen echter hoog oplopen (tot 40 punten), ze kunnen ook ten nadele van performaal zijn (Cotton et al., 2005) en ze worden vooral gevonden bij jongere kinderen: bij de meeste adolescenten is het verschil tussen Verbaal en Performaal IQ verdwenen (Cotton et al., 2005). Laatstgenoemde bevinding heeft betekenis voor de interpretatie van de oorzaak van het verschil.
Taal en andere cognitieve functies
Jongens met Duchenne spierdystrofie behalen bij testonderzoek in de meeste opzichten nauwelijks lagere scores dan leeftijdgenoten in de algemene bevolking. Maar op het tussenveld van taal en geheugen lijken zij wat minder sterk. Dat blijkt bijvoorbeeld bij opdrachten waarin het kind onmiddellijk na mondelinge aanbieding reeksen cijfers of woorden moet nazeggen of verhalen moet navertellen. Anders gezegd, de verbale spanne (het maximale aantal elementen dat na eenmalige aanbieding kan worden gereproduceerd) is vaak wat geringer dan past bij de normwaarden van de test (Hinton et al., 2007). In de leeftijd tussen zes en tien jaar lijken jongens met Duchenne spierdystrofie minder werkwoorden te gebruiken en zinnen vaker niet af te maken dan normaal ontwikkelende jongens van dezelfde leeftijd (Marini et al., 2007), maar dat kan samenhangen met verschil in mentale ontwikkeling. Deze kinderen hadden zowel receptief (begrijpen) als productief (praten) een normale taalstructuur (ENCYCL-Taalverwerving). Geheugen en leren zouden als zodanig passen bij de intelligentie en dus niet selectief gestoord zijn.
Schoolvaardigheden
Duchenne jongens behalen, ook als zij een gemiddelde of bovengemiddelde intelligentie hebben, bij onderzoek van schoolvaardigheden globaal zwakkere scores dan gezonde broertjes en zusjes met dezelfde woordenschat en dezelfde thuissituatie. Er zijn Duchenne-jongens die woorden en psudowoorden minder snel harop lezen dan op grond van leeftijd en intelligentienivesu verwacht kon worden (Hendriksen et al., 2006). Achterblijven in rekenen valt vooral op (Hinton et al., 2004). Schoolvorderingen hangen niet alleen af van de intelligentie, maar ook van een reeks andere factoren, variërend van lichamelijke toestand en genetische achtergrond tot schoolverzuim, emotioneel welbevinden en acceptatie door klasgenoten.
Gedrag
Aandachtstekort met hyperactiviteit (ADHD), stoornissen in het autismespectrum en obsessief-compulsieve stoornissen lijken bij Duchenne spierdystrofie iets meer voor te komen dan in de algemene bevolking (Hendriksen & Vles, 2008). Verschijnselen van de twee eerstgenoemde gedragsafwijkingen doen zich vaak zich voor bij zwakzinnige kinderen. Of dat verband ook bestaat bij jongens met Duchenne spierdystrofie is nog niet onderzocht.
fajenn27 januari, 2011 - 12:37
Psychosociaal functioneren
Psychosociaal functioneren
De levensweg van een jongen met Duchenne spierdystrofie verloopt van vrijwel normaal functioneren in het eerste levensjaar naar ernstige spierzwakte en afhankelijkheid van zorgverlening in de adolescentie en ouder.
Voorschoolse periode
• Door hun slechte lopen verkeren Duchenne jongens vanaf de peuter- en kleuterjaren in een uitzonderingspositie met risico op gepest worden en sociaal isolement.
• Tussen 4 en 8 jaar wordt veelal begonnen met prednisonbehandeling. Vollemaansgezicht en toename van lichaamsgewicht behoren tot de bijwerkingen, die beide een zeer negatief effect hebben op de uiterlijke verschijning van een kind en maar al te gemakkelijk kunnen leiden tot schaamte en gebrek aan zelfvertrouwen.
• Veel ouders zijn geneigd hun zwaar belaste kind te ontzien. Opvoeding tot positief sociaal en verantwoordelijk gedrag is te verkiezen, zowel voor het kind zelf als voor zijn omgang met de omgeving.
Schooljaren, voorbereiding op tweede en derde levensdecennium
• De keuze van de school vormt een beslissend moment. Een aanmerkelijke minderheid van de jongens met Duchenne spierdystrofie is immers zwakbegaafd of zwakzinnig. Kinderen met lichamelijke functiebeperkingen hebben, nog meer dan andere kinderen, af en toe succes nodig ter sterking van hun zelfvertrouwen, ook in hun schoolprestaties. Rekening moet worden gehouden met te verwachten extra schoolverzuim in verband met veelvuldige medische controles. Fysiotherapeutische behandeling ter voorkoming van contracturen wordt zo mogelijk ingepast in het schooldagprogramma.
• Vaardigheid in het omgaan met de persoonlijke computer en verwante apparatuur is voor de toekomst van kinderen met Duchenne spierdystrofie van groot belang. De computerwereld is overwegend Engelstalig, onderwijs in de Engelse taal behoeft mede daarom extra aandacht.
• Duchenne jongens kunnen zich richten op passende vormen van sport zoals rolstoelhockey, denksporten als schaken of bridgen, computerspellen. Voor muziekbeoefening komen muziekinstrumenten in aanmerking die beperkte handvaardigheid vergen.
Thuis blijven of zelfstandig wonen
Adolescenten en jongvolwassenen verlaten in onze samenleving het ouderlijk huis. Voor die keuze staan ook adolescenten met Duchenne spierdystrofie die opteren voor tracheostomale ademhalingsondersteuning (zie onder Lichamelijke verschijnselen, de paragrafen over Ademhalingsondersteuning en Levensduur).
• In het ouderlijk huis blijven wonen betekent over het algemeen kiezen voor zekerheid en bekende, vertrouwde zorg. Nadelen zijn: veelal weinig contact met leeftijdsgenoten, voortzetting van de afhankelijkheidsband met de ouders en daardoor dikwijls onvoldoende autonomie, en niet te vergeten toenemend zware belasting van de zorgverlenende ouders, meestal vooral de moeder.
• Zelfstandig wonen, bijvoorbeeld in een Fokusproject of in Het Dorp vermindert het risico van sociaal isolement, vergroot de autonomie, en kan anderzijds meer risico geven op onvoldoende continuïteit van de zorg. Voor sommige jongeren met zwakke intellectuele begaafdheid zijn wellicht meer beschutte woonvormen nodig. Zelfstandig wonen stelt aan zwakzinnigen te hoge eisen.
fajenn27 januari, 2011 - 12:41
Begeleiding
Begeleiding
Jongens met Duchenne spierdystrofie hebben bij voortduring medische en paramedische zorg nodig waarbij veel verschillende specialismen betrokken zijn. Van belang is dat één van de medische behandelaars functioneert als vaste coördinator. Tot aan de fase van ademhalingsondersteuning is dat vaak de behandelende revalidatiearts en daarna soms de thuisbeademingsarts. Neuropsychologisch onderzoek is zinvol als een probleem daar aanleiding toe geeft, zoals bij vertraagde taalverwerving, of op beslissingsmomenten, bijvoorbeeld voor het begin van de basisschool (Bushby et al., 2010). Bij de beslissing over een woonvorm kan in voorkomende gevallen neuropsychologische diagnostiek ook van belang zijn. Met de fijne handmotoriek moet bij het onderzoek rekening worden gehouden om eventuele nadelen in de uitvoering van performale tests en schrijfwerk te kunnen verdisconteren.
fajenn27 januari, 2011 - 12:43
Fragmenten uit interviews met T, zijn moeder en raadsman
Fragmenten uit interviews met T, zijn moeder en raadsman
T : Mijn broer en ik kregen al heel jong de eerste tekenen van spierdystrofie. Van de eerste jaren herinner ik mij alleen dat het lopen steeds moeilijker werd. Op school werden mijn broer en ik regelmatig geplaagd. Een keer hadden jongens van de school mijn broer in de heg geduwd en vreselijk moeten lachen omdat hij er niet uit kon komen.
Moeder: T. had nooit veel belangstelling voor de boerderij. Hij hield van studeren en had iets ondernemends. Omdat wij nog twee andere gehandicapte kinderen hadden is T al jong in de Johanna Stichting opgenomen. We bedongen wel dat hij elk weekeinde thuis kon komen.
T.: In de Johanna Stichting werd nog veel meer geplaagd dan thuis op school. Het vroeg uit huis zijn was toch goed voor mij geloof ik. Het maakte me zelfstandig en bezorgde me vrienden. ….Ik speelde met andere spierzieke jongens en meisjes rolstoelhockey. De revalidatieartsen waren daarop tegen omdat ze meenden dat de spierinspanning slecht was voor mensen met een spierziekte maar dat weerhield ons niet. ‘Dan maar een beetje minder leven’ zeiden we tegen elkaar.
T.: Ik ben als een van de eerste spierzieken in Nederland chronisch beademd, ongeveer vanaf mijn 23ste jaar. De keuze tussen wel of niet beademd worden was voor mij geen probleem. Leven is nu eenmaal beter dan niet leven. Ik heb er nooit spijt van gehad. Nu vergeet ik [de beademing] meestal. (…) Zitten is niet gemakkelijk voor me, ondanks mijn korset. Ook in bed moet ik vaak de goede positie zoeken om geen pijn te hebben. Eigenlijk voel ik me vaak niet prettig. Gelukkig kan ik mijn duim nog bewegen en daarmee mijn [computer] paneel bedienen.
T.: Ik heb een tijd lang met een vriendin samengewoond. Zij was ook gehandicapt maar minder dan ik. Zij is weggegaan omdat ik zoveel zorg nodig had. Dat gaf haar weinig privacy.
T.: ’s Middags heb ik tijd om dingen te doen. Soms heb ik een afspraak met andere mensen of ik kan naar de winkel gaan om inkopen te doen. Ik kan gaan lezen of iets doen op mijn computer, of tv kijken of telefoneren. Bijna elk jaar volg ik een schriftelijke cursus. (…) Ik wil nog graag een beetje doorleven, er is nog zoveel dat me interesseert.
Raadsman: [Over seksbehoefte] in zijn algemeenheid kan ik …alleen maar zeggen dat het terdege een probleem is. Het is geen onderwerp waarover ze (de spierzieken) gemakkelijk praten. Zeker is dat ze op het internet gemakkelijk de weg vinden naar erotische sites. Wat in dit opzicht [hier] wordt gedaan is mij niet bekend. Het taboe op seks is nog niet helemaal weg.
(zie voor de gehele interviews: Jennekens en Kater, 2004)
fajenn27 januari, 2011 - 12:49
Literatuur
Literatuur
Anderson JL, Head SI, Rae C, Morley JW (2002) Brain function in Duchenne muscular dystrophy. Brain 125: 4-13
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomeszko J, Constantin C for the DMD Care Considerations Working Group (2010) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurology 9: 77-93
Cotton S, Voudouris NJ, Greenwood KM (2001) Intelligence and Duchenne muscular dystrophy: Full-Scale, Verbal, and Performance intelligence quotients. Developmental Medicine & Child Neurology 43: 497-501
Cotton SM, Voudouris NJ, Greenwood KM (2005) Association between intellectual functioning and age in children and young adults with Duchenne muscular dystrophy: further results from a meta-annalysis. Developmental Medicine & Child Neurology 47: 252-265
Cyrulnik SE, Fee RJ, de Vivo DC, Goldstein E, Hinton VJ (2007) Delayed developmental language milestones in children with Duchenne’s muscular dystrophy. Journal of Pediatrics 150; 474-478
De Die-Smulders CEM, Faber CGM, Pinto Y, Schrander-Stumpel CTRM (2004) Klinische genetica: Duchenne spierdystrofie. Patient Care 31: 402-408
Dreyer PS, Steffensen BF, Pedersen BD (2010) Life with home mechanical ventilation for young men with Duchenne muscular dystrophy. Journal of Advanced Nursing 66: 753-762
Hendriksen JCM, Vles JSH (2006) Are males with Duchenne muscular dystrophy at risk for reading disability? Pediatric Neurology 34: 296-300
Hendriksen JGM, Vles JSH (2008) Neuropsychiatric disorders in males with Duchenne muscular dystrophy: frequency rate of attention deficit hyperactivity disorder (ADHD), autism spectrum disorder and obsessive-compusive disorder. Journal of Child Neurology 23: 477-481
Hinton VJ, Fee RJ, Goldstein EM, DeVivo DC (2007) Verbal and memory skills in males with Duchenne muscular dystrophy. Developmental Medicine & Child Neurology 49: 123 – 128
Hinton VJ, DeVivo DC, Fee R, Goldstein E, Stern Y (2004) Investigation of poor academic achievement in children with Duchenne muscular dystrophy. Learning Disability Research Practice 19: 146- 154
Jennekens FGI, Kater L (2004) Wie zwak is moet sterk zijn. De recente geschiedenis van spierzieken. Baarn: TIRION pp 63-79
Marini A, Lorusso ML, D’Angelo MG, Civati F, Turconi AC, Fabbro F, Bresolin N (2007) Evaluation of narrative abilities in patients suffering from Duchenne muscular dysrophy. Brain & Language 102: 1-12
Meinesz AF, Bladder G, Goorhuis JF, Fock JM, Staal-Schreinemachers AL, Zijlstra JG, Wijkstra PJ (2007) 18 jaar ervaring met chronische beademing bij patiënten met spierdystrofie van Duchenne. Nederlands Tijdschrift voor Geneeskunde 151: 1830-1833
Moxley RT, Pandya S, Ciafaloni E, Fox DJ, Campbell K (2010) Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. Journal of Child Neurology 25: 1116-1129
Ricotti V, Roberts RG, Muntoni F (2011) Dystrophin and the brain. Developmental Medicine & Child Neurology 53:12
Taylor PJ, Betts GA, Maroulis S, Gilissen C, Pedersen RL, Mowat DR, Johnston HM, Buckley MF (2010) Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS ONE 5: e8803
Van Essen J, Busch HF, te Meerman GJ, ten Kate LP (1992) Birth and population prevalence of Duchenne muscular dystrophy in the Netherlands. Human Genetics 88: 258-266
fajenn27 januari, 2011 - 12:51
Neurofibromatose type 1
Neurofibromatose type 1fajenn12 april, 2011 - 21:02
Inleiding
Inleiding
Neurofibromatosis 1 (NF1) is een erfelijke ziekte van huid en zenuwstelsel en van verschillende andere organen. “Neuro” in de naam van de ziekte verwijst naar de aandoening van het zenuwstelsel en “ fibromatosis” naar bindweefselachtige gezwellen. De gezwellen zijn vezelig en celrijk.Beide kenmerken kunnen per gezwel variëren. Een aanmerkelijke minderheid van de kinderen heeft cognitieve stoornissen, de meerderheid heeft leerproblemen en psychosociale problemen.
Verschijnselen van enkele andere ziekten overlappen met die van NF1 (Boyd et al., 2009). Neurofibromatosis 2 veroorzaakt tumoren (Schwannomen, meningeomen, ependymomen) in het centrale zenuwstelsel (CZS) maar leidt niet tot cognitieve stoornissen. De verschijnselen van het perifere zenuwstelsel staan minder op de voorgrond dan bij NF1. In vergelijking tot NF1 zijn de huidafwijkingen gering.
fajenn12 april, 2011 - 21:03
Andere teksten op internet
Andere teksten op internet
Op het internet zijn een groot aantal andere veelal Engelstalige websites over deze ziekte beschikbaar. www.huidziekten.nl/ en www.nvk.nl zijn websites voor dermatologen en kindergeneeskundigen. Onder “richtlijnen” is in deze websites de “NVK –leidraad voor de medische begeleiding van kinderen met neurofibromatosis type 1” opgenomen. De leidraad dateert van 2006. www.neurofibromatose.nl is de website van de Neurofibromatose Vereniging Nederland. Via deze site kan de brochure “ Leer-en gedragsstoornissen bij kinderen met neurofibromatose type 1” worden opgezocht. De brochure is geschreven door leden van het NF1 Expertisecentrum van Erasmus MC-Sophia Kinderziekenhuis Rotterdam. De brochure bevat veel praktisch belangrijke adviezen. De site dateert van 2009. www.kinderneurologie.eu : de tekst over neurofibromatose type 1 op deze website is geschreven door een kinderneuroloog en bevat veel deskundige informatie. De tekst is vooral bestemd voor niet-beroepshalve geïnteresseerden en dateert van 2007. www.ncbi.nih.gov/omim/162200 is een website van de John Hopkins University over monogenetische ziekten waarin uitvoerig aandacht wordt besteed aan NF1. OMIM is de afkorting van “ Online Mendelian Inheritance in Man”
fajenn14 april, 2011 - 12:02
Epidemiologie
Epidemiologie
Op NF1 wijzende verschijnselen werden in Duitsland vastgesteld bij tenminste één per 3000 kinderen van 6 jaar, ofwel 33 per 100.000 kinderen (Lammert et al., 2005). In Noord Ierland is voor kinderen jonger dan 16 jaar een lager getal opgegeven (McKeever et al., 2008).
fajenn31 mei, 2011 - 19:02
Erfelijkheid en genmutaties
Erfelijkheid en genmutaties
Erfelijkheid
De ziekte ontstaat in ongeveer de helft van alle gevallen spontaan. Dat wil zeggen dat geen enkel familielid NF1 heeft. Ieder kind van een ouder met NF1 heeft een kans van 50% op dezelfde gen-afwijking. De penetrantie (percentage gendragers met ziekteverschijnselen) is op 20-jarige leeftijd vrijwel 100%.
Genmutatie
De ziekte is het gevolg van een mutatie in een gen op de lange arm van chromosoom 17 (q 11.2). De kans op een spontane mutatie in dit gen is groot. De mutaties verschillen van volledige afwezigheid van het gen bij een microdeletie tot een puntmutatie in het gen. Bij microdeleties zijn de NF1 verschijnselen ernstiger dan bij mutaties in het NF1-gen (Pasmant et al., 2011).
De ziekteverschijnselen verschillen sterk tussen en zelfs binnen families met NF1 (Boyd et al., 2009). Zelfs de verschijnselen die het gevolg zijn van één en dezelfde mutatie variëren. De variabiliteit berust althans ten dele op invloed van andere genen die niet met het NF1-gen verbonden zijn (“genetic modifiers”) (Sabbagh et al., 2009).
Het gen bevat de code voor het eiwit neurofibromine. Dit eiwit is in veel cellen aanwezig, onder andere in neuronen en gliacellen. Neurofibromine gaat het ontstaan van tumoren tegen (Boyd et al., 2009).
Mozaïcisme
Het NF1-gen kan in sommige cellen van een kind afwijkend zijn en in andere cellen niet. Er zijn dan twee cellijnen: de ene met cellen die allemaal het afwijkende gen bevatten, de tweede zonder afwijkend NF1-gen. Dit wordt mozaïcisme genoemd. Het kan leiden tot ongewone beelden, bijvoorbeeld alleen NF1-verschijnselen aan een arm of been (segmentale NF1), en tot diagnostische problemen (Boyd et al., 2009).
fajenn31 mei, 2011 - 19:05
Hersenafwijkingen
Hersenafwijkingen
T2 gewogen MRI’s van de hersenen tonen bij vrijwel alle kinderen met NF1 pleksgewijze kleine ophelderingen (“unidentified bright objects”, UBO’s), vooral in de witte stof van de grote hersenen, maar ook in het gebied van de basale kernen, thalamus, hersenstam en cerebellum.(ENCYCL-Anatomie van de hersenen). Mogelijk is op deze plaatsen de microstructuur van het hersenweefsel nog meer dan elders abnormaal, door een teveel aan vocht of door holtes (vacuoles) in het myeline (van Engelen et al., 2008). De UBO’s worden kleiner en verdwijnen bij het ouder worden maar niet overal of niet overal in dezelfde mate.Figuur 1 Een "unidentified bright object".
Overgenomen via Google afbeeldingen van www.drugaware.com.au
Bij ongeveer 50% van de kinderen met NF1 is de hoofdomtrek te groot (macrocefalie) als gevolg van een te grote hersenmassa (megalencefalie). De ongewone grootte van de hersenen kan niet eenvoudig worden toegeschreven aan teveel grijze en/of te veel witte stof.
Een opvallende afwijking is het grote volume van de belangrijkste verbinding tussen de linker- en rechterhersenhelft; deze verbinding heet “balk” of corpus callosum (ENCYCL-Anatomie van de hersenen).
Over de betekenis van deze afwijkingen voor de cognitie is veel gespeculeerd en onderzoek verricht, maar een definitieve conclusie ontbreekt (Payne et al., 2010).
fajenn31 mei, 2011 - 20:58
Lichamelijke verschijnselen, medische controle
Lichamelijke verschijnselen, medische controle
Verschijnselen van belang voor de diagnose
Voor de diagnose zijn zeven criteria van belang; de diagnose is “zeer waarschijnlijk” als bij een kind twee van deze criteria worden aangetroffen (DeBella et al., 2000). De criteria zijn:
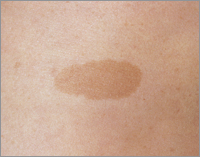
1. Café-au-lait (CAL) vlekken op de huid: tenminste zes. De vlekken nemen in de kinderjaren toe in aantal en grootte. Ze zijn niet aanwezig op behaarde hoofd, wenkbrauwen, handpalmen en voetzolen.
Figuur 2. Een café au lait vlek.
Overgenomen via Google afbeeldingen van www.kiesbeter.nl
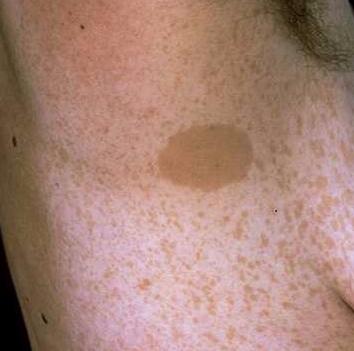
2. Sproetvorming in oksel of lies: de zogeheten sproeten ("freckels") zijn kleiner en donkerder dan CAL-vlekken. Op zesjarige leeftijd aanwezig bij 60-90% van de kinderen met NF1.
Figuur 3. Sproetvorming:
Overgenomen via Google afbeeldingen van www.mrcopht.cm. Ongeveer in het midden is een CAL-vlek te zien, daarom heen de sproetvorming.
3. Lisch noduli: tenminste twee lichtbruine plekjes (celophopingen) in de iris; de plekjes bevatten melanine. Ze worden bij 40% van de zesjarige kinderen met NF1 gevonden en zijn uiteindelijk bij alle NF1 patiënten aanwezig.
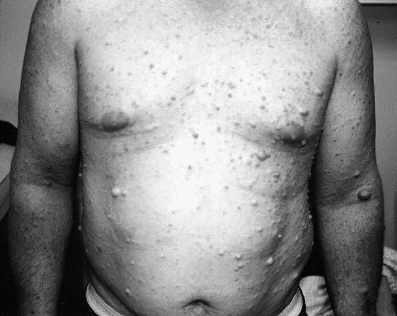
4. Neurofibromen in of onder de huid: tot één of enkele centimeters grote, zachte, roodblauwige tumoren uitgaande van zenuwen in of onder de huid. Ze kunnen al voor de puberteit aanwezig zijn en ze nemen daarna in aantal toe. Op de leeftijd van 20 jaar hebben bijna alle NF1 patiënten neurofibromen. Plexiforme neurofibromen zijn netwerken van tumorweefsel, die zich in of onder de huid of in het inwendige van het lichaam – bijvoorbeeld in de oogkas, de halsstreek of de borstholte – als varens uitbreiden. Op de leeftijd van 18 jaar heeft een kwart van de NF1 jongeren een dergelijke tumor.
Figuur 4. Neurofibromen bij een jong volwassenen:
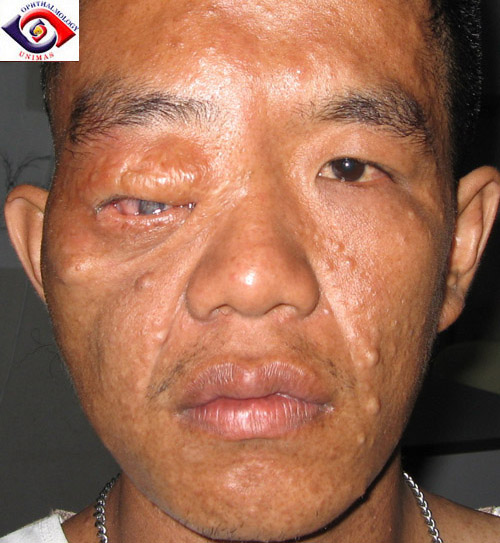
Overgenomen via Google afbeeldingen van www.nfinfo.nlFiguur 5a. Plexiform neurofibroom in de rechter oogkas uitpuilend onder bovenste ooglid. Neurofibromen in het gelaat:
Overgenomen via Google afbeeldingen van www.sarawakeyecare.comFiguur 5b. Plexiform neuribroom in de linker oogkas bij een jong kind:
Overgenomen via Google afbeeldingen van www.us.oup.com. Plexiforme neurofibromen groeien vooral in de kinderjaren. Er zijn dan als regel nog geen neurofibromen in het gelaat
5. Gliomen in de nervus opticus. Tumoren uitgaande van astroglia cellen in de gezichtszenuw (nervus opticus). Bij ongeveer een zesde van alle NF1 kinderen. De tumoren geven niet altijd klachten.
6. Skeletafwijkingen: scoliose, verkromming van pijpbeenderen, soms een schijngewricht, abnormale ontwikkeling van schedelbeenderen.
7. In 50% van de gevallen heeft één van de ouders NF1.
Andere verschijnselen
Een groot aantal andere ziekteverschijnselen kan bij kinderen met NF1 ontstaan. Ze worden hier genoemd om aan te geven hoe veelvormig de ziekte kan zijn (Huson & Korf, 2002): te geringe lichaamslengte (bij 30% van de kinderen); te grote hersenmassa (macrocefalie, bij 45%); epilepsie; hydrocefalus door aqueductstenose; te hoge oogboldruk; afwijkende stem; meningokèle op borsthoogte; goedaardige gezwellen (sommige hersentumoren, neurofibromen uitgaande van maag-darmwand), tumoren met hormonale effecten (de neuro-endocriene tumoren zoals carcinoïd en feochromocytoom); kwaadaardige tumoren zoals de sarcomen vooral uitgaande van plexiforme neurofibromen en de tumoren uitgaande van spierweefsel; woekering van beenmergcellen (myeloïde leukemie); te vroege of late puberteit; afwijkingen van het vaatstelsel (moyamoya-syndroom); stenose van de nierarterie, hypertensie.
Graden van ernst van de ziekte
NF1 is (1) minimaal als er alleen enige huidafwijkingen zijn, (2) licht als de huidafwijkingen zichtbaar zijn, (3) matig ernstig als de verschijnselen bedreigend voor de gezondheid maar wel hanteerbaar zijn, (4) ernstig als de verschijnselen zeer bedreigend en moeilijk hanteerbaar zijn (zie Jennekens-Schinkel & Jennekens, 2008, pp 503-504).
Medische begeleiding en behandeling van kinderen
NF1 kan talrijke ziekteverschijnselen veroorzaken, maar bij afzonderlijke kinderen ontbreken vele ervan en voor zover wel aanwezig behoeven ze niet ernstig te zijn. Jaarlijkse medische controle is noodzakelijk. Volgens een internationale richtlijn dienen daarbij de volgende punten aan de orde te komen (Ferner et al., 2007):
Zijn er problemen op school?
Oogheelkundig onderzoek door oogarts (tot 10de jaar, volgens NVK richtlijn, zie www.nvk.nl )
Hoofdomtrek en lichaamslengte: is het hoofd te groot en de lichaamslengte onvoldoende?
Is puberteit te vroeg of te laat?
Is bloeddruk te hoog?
Hart- en vaatonderzoek: zijn er aangeboren afwijkingen?
Wervelkolom inspectie: is er scoliose?
Zijn de huidafwijkingen toegenomen, zijn ze zichtbaar?
Is nader onderzoek voor specifieke klachten nodig?
fajenn31 mei, 2011 - 21:03
Cognitief functioneren
Cognitief functioneren
Er kunnen verschillende redenen zijn voor het vaststellen van het cognitieve functioneren van kinderen met NF1, bijvoorbeeld tegenvallend leren op school, keuze van gewoon of speciaal onderwijs, of ongewenst/onbegrepen gedrag.
Bij neuro-, ontwikkelings- of schoolpsychologisch onderzoek moet rekening gehouden worden met verschijnselen die de resultaten van testonderzoek nadelig kunnen beïnvloeden (zie Jennekens-Schinkel & Jennekens, 2008, pp 493-494). Hier worden genoemd:
Bij 10% van de peuters en kleuters met NF1 loopt de motorische ontwikkeling achter op de – normale – mentale ontwikkeling: van belang omdat de tests op deze leeftijden motorische reactie vergen
In de schoolleeftijd is 50% van de kinderen met NF1 motorisch onhandig, zoals blijkt bij bijvoorbeeld kralen rijgen of trommelen met de vingers, en een kwart van de kinderen is motorisch traag: van belang omdat leren schrijven en tekenen motoriek vergen
Visusstoornissen. Controle voorafgaande op testonderzoek is nodig
Intelligentie
Het intelligentiequotiënt (IQ) is bij schoolkinderen met NF1 gemiddeld ongeveer een standaardeviatie lager dan gemiddeld in de algemene bevolking: het Totale IQ (TIQ) is bij kinderen met NF1 gemiddeld ongeveer 86. De spreiding van de IQ’s is normaal; dit betekent dat ongeveer de helft van de kinderen een TIQ lager dan 85 heeft, wat kan wijzen op zwakbegaafdheid of zwakzinnigheid. Vier tot achttien procent van de kinderen met NF1 heeft IQ’s lager dan 70, passend bij zwakzinnigheid (Krab et al., 2008). Maar hoogbegaafde NF1 kinderen zijn er ook (zie Jennekens-Schinkel & Jennekens, 2008, p 494).
Het Verbale IQ is dikwijls hoger dan het Performale IQ; dit is echter niet kenmerkend voor de intelligentie bij NF1.
Duidelijke aanwijzingen dat de intelligentie als regel daalt bij het ouder worden ontbreken.
Kinderen met minimale NF1 presteren in neuropsychologisch testonderzoek beter dan kinderen met ernstiger NF1 (Krab et al., 2008).
Welke veranderingen in de hersenen oorzaak zijn van de mindere intelligentie is tot nu toe niet met zekerheid vastgesteld (Payne et al., 2010).
Visuospatiële functies
Het zien (visus) is bij verwerken van ruimtelijke (spatiële) informatie in het dagelijks leven maar zeker ook bij testonderzoek erg belangrijk, vandaar de term visuospatiële functies. In de NF1 literatuur gelden deze functies als gestoord. Deze opvatting is vooral gebaseerd op onderzoek met behulp van de “Lijnoriëntatietest”, in Nederland ook wel genoemd hellingsdetectietest. In deze test moet de onderzochte de helling van telkens twee lijnen vergelijken. Als groep doen kinderen met NF1 dit minder goed dan kinderen zonder NF1 (zie Jennekens-Schinkel & Jennekens, 2008, pp 495-497).
De praktische betekenis van een dergelijk tekort staat nog niet vast. Als groep zijn kinderen met NF1 ook minder sterk dan gezonde leeftijdgenootjes in natekenen van figuren, klaarblijkelijk ten dele als gevolg van een visuospatiële stoornis en ten dele als gevolg van onvoldoende fijnmotorische coõrdinatie (Krab et al., 2011).
Geheugen en leren
In groepen kinderen met NF1 variëren de gemiddelde scores voor geheugen en leren van “passend bij het gemiddelde IQ” tot “wat beter dan het gemiddelde IQ zou doen verwachten” (zie Jennekens-Schinkel & Jennekens, 2008, p 497). Motorisch leren is bij de meeste kinderen met NF1 niet gestoord. Hierin behoeft de oorzaak van de moeite met (na)tekenen, leren schrijven en andere handelingen waarin “construeren” een rol speelt, niet te worden gezocht (Krab et al., 2011).
Taal en spraak
Taalverwerving: een minderheid van NF1 kinderen en adolescenten is traag in taalverwerving, zowel in het zich uitdrukken in taal (taalexpressie) als in het begrijpen (taalreceptie) (zie ENCYCL-Taalverwerving). Gemiddeld over groepen kinderen met NF1 zijn mondeling uitdrukken, luisterbegrip en verbaal abstractievermogen gemiddeld laag normaal (Thompson et al., 2010; Hyman et al., 2005; zie Jennekens-Schinkel & Jennekens, 2008 voor een overzicht, p 498).
Spraakproblemen: de spraak ontwikkelt trager dan normaal (Thompson et al., 2010). Bovendien hebben veel kinderen met NF1 een hese of nasale stem of een stem waarvan de klankstructuur gestoord is. Ook heeft de stem bij velen een beperkt toonhoogtebereik en beperkte toonhoogteregulatie. Daardoor klinkt de spraak monotoon, wat soms ten onrechte lijkt op emotionele vlakheid van het kind. Veel kinderen hebben ook moeite met articuleren van medeklinkercombinaties waardoor ze slecht verstaanbaar zijn. De stem- en spraakafwijkingen zijn in variabele mate aanwezig, bij sommige kinderen helemaal niet. Ze komen meer voor bij kinderen dan bij volwassenen met NF1 (Alivuotila et al., 2010). Hoewel de spraak tekort kan schieten in vloeiendheid, stotteren de kinderen niet. De complexe oorzaken van de spraakproblemen zijn niet ontrafeld.
Aandacht en executief functioneren
Aandacht, zich richten op een onderwerp of een handeling en die gerichtheid volhouden, wordt wel onderscheiden van executieve functies. Onder de laatste verstaat men uiteenlopende cognitieve en gedragsmatige vaardigheden die doelgericht handelen mogelijk maken, zoals: een plan maken, organiseren, flexibiliteit van denken, abstracte begripsvorming en impulsbeheersing. Tests van de kinderen leveren voor aandacht en executieve functies vaak gegevens op die niet of weinig overeenkomen met de beoordelingen door ouders en leerkrachten. In een periode van acht jaren werden bij 199 kinderen met NF1 en 55 gezonde broertjes of zusjes van de patiëntjes zowel tests als vragenlijsten gebruikt om meer inzicht te krijgen in aandacht en executieve functies. De kinderen varieerden in leeftijd van 6 tot 16 jaar. De vragenlijsten betreffende aandacht (Conners Attention Deficit Scale) en executief functioneren (BRIEF: Behavior Rating Inventory of Executive Function) werden ingevuld door ouders en leerkrachten.
1. Een grote minderheid van de kinderen met NF1 (40%) leed volgens ouders in het dagelijkse bestaan aan een aandachtstekort al of niet met overmatige beweeglijkheid (attention deficit with hyperactivity disorder, ADHD of attention deficit disorder, ADD). Alleen hyperactiviteit kwam bij kinderen met NF1 aanzienlijk minder voor. De leerkrachten zagen ook meer onaandachtig handelen al of niet met hyperactiviteit bij de kinderen met NF1, maar de percentages waren wat lager dan in de beoordelingen door ouders (mogelijk doordat de situaties thuis en op school verschillen, of door verschil in kijk van de beoordelaars).
2. Volgens ouders had bijna 30% van de kinderen met NF1 tekorten in executief functioneren die van klinische betekenis waren (dat wil zeggen 1,5 standaarddeviatie meer dan normaal ontwikkelende controlekinderen). De tekorten werden gezien in alle dimensies die de BRIEF inventariseert.
3. Het beeld werd niet anders wanneer rekening gehouden werd met het niveau van de verbale intelligentie (bepaald met behulp van de subtest Begrijpen van de WISC).
4. De resultaten van de bij de kinderen zelf afgenomen tests betreffende aandacht en executieve functies correleerden maar ten dele met de gegevens van ouders en leerkrachten. Tests lijken gevoeliger voor tekorten in werkgeheugen, aandacht en organisatievaardigheden, terwijl de vragenlijsten zoals de BRIEF veeleer wijzen op tekorten in impulsbeheersing en sociaal functioneren. In elk geval informeren de vragenlijsten over tekorten in het dagelijks gedrag terwijl de tests misschien meer tonen wat het kind onder ideale omstandigheden kan of niet kan (Payne et al., 2011).
fajenn31 mei, 2011 - 21:24
Schoolvaardigheden
Schoolvaardigheden
Spellen, technisch lezen, begrijpend lezen, rekenen
Rotterdams onderzoek van schoolvaardigheden maakte gebruik van de leerlingvolgsystemen voor de vakken spellen, technisch lezen, begrijpend lezen en rekenen. Alleen kinderen (leeftijd 7–17 jaar) met voldoende gehoor en gezichtsvermogen en zonder aantoonbare hersenaandoeningen (zoals epilepsie) werden bij het onderzoek betrokken. Ofschoon de gebruikte maat, het “didactische leeftijdsequivalent” bezwaren heeft (Evers & Resing, 2007), bleek duidelijk dat meer dan 60% van de kinderen met NF1 op school moeilijk vordert in deze vakken. De beperkingen kwamen in de vier vakken ongeveer even vaak voor, er was dus niet één vak extra moeilijk. De leerproblemen werden specifiek genoemd als de kinderen beperking hadden in een of meer vak(ken) bij een IQ hoger dan 84. Ze werden algemeen genoemd als het IQ lager dan 85 was. Specifieke en algemene leerproblemen kwamen ongeveer even vaak (elk bij bijna 40% van de kinderen) voor (Krab et al., 2008; zie voor literatuuroverzicht Jennekens-Schinkel & Jennekens, 2008, pp 500-501). Maar dit betekent ook dat de kinderen onderling sterk kunnen verschillen, zoals geïllustreerd door het volgende voorbeeld. Van 17 Belgische kinderen met NF1 hadden 8 kinderen leerproblemen. Vier kinderen hadden alleen problemen met taal, één kind had alleen zeer ernstige problemen met rekenen en drie kinderen hadden taal- en rekenproblemen. “Begrijpend lezen” was bij alle 8 kinderen in orde (Descheemaeker et al., 2005).
Het handschrift
Veel kinderen met NF1 hebben, in vergelijking met normaal ontwikkelende leeftijdgenoten van hetzelfde geslacht, een rommelig aandoend handschrift: meer letters onder of boven de lijn, wisselvalliger lettergrootte en wisselvalliger onderlinge afstanden tussen letters en woorden (Gilboa et al., 2010). Van alle kinderen was het IQ “hoger dan 70”, maar deze omstandigheid sluit niet uit dat toch verschillen in cognitief niveau terug te vinden waren in het schrijfwerk. Immers, van een opstelletje was de inhoud – ideeën, woordkeuze, vloeiendheid van zinnen, organisatie van het schrijven – gemiddeld zwakker dan bij kinderen zonder NF1 (Gilboa et al., 2010).
Oorzaken van leerproblemen
Het is moeilijk te zeggen of de leerproblemen begrepen kunnen worden uit cognitieve functiestoornissen. Wel is samenhang gevonden tussen verhoudingsgewijs laag verbaal IQ en vooral bij jongens specifieke tekorten in lezen, spellen en rekenen. Onvoldoende vasthouden of wisselen van aandacht, tekortschieten in planning van taken, en moeilijkheden bij het verwerken van ruimtelijke relaties hielden ook verband met de leerproblemen (Hyman et al., 2005). Slaapproblemen, met name nachtelijke angst en slaapwandelen, komen bij ruim één op de drie kinderen met NF1 veel voor (Johnson et al., 2005); ook deze zouden een factor kunnen zijn in het ontstaan van de leerproblemen.
fajenn31 mei, 2011 - 21:33
Schoolloopbaan
Schoolloopbaan
Rotterdams onderzoek wees uit dat slechts 10% van de kinderen tot het moment van onderzoek een normale schoolloopbaan had doorlopen. De overgrote meerderheid van de kinderen met NF1 (73%) ontving extra hulp bij het schoolse leren, vaak in combinatie met ondersteuning van de motorische ontwikkeling of met logopedie. Een grote minderheid (38%) volgde speciaal onderwijs, waarvan 5% scholen voor zeer moeilijk lerende kinderen. Ruim een derde (36%) van de kinderen met NF1 op scholen voor regulier basisonderwijs doubleerde een leerjaar (ruim twee keer zoveel als in de algemene schoolpopulatie, waar 16% doubleerde) (Krab et al., 2008).
fajenn31 mei, 2011 - 21:35
Psychosociaal functioneren
Psychosociaal functioneren
De sociale vaardigheden van NF1 kinderen schieten naar het oordeel van ouders en leerkrachten niet tekort. Toch heeft NF1 vaak gevolgen in het sociale verkeer, bijvoorbeeld door de zichtbaarheid van NF1 afwijkingen. Vooral in de puberteit ontstaan hierdoor problemen. De jongeren proberen de afwijkingen te verhullen, soms mijden ze sport en spel omdat hals en delen van de romp daarbij zichtbaar zijn, of ze ontlopen sociale contacten. Kinderen met zichtbare afwijkingen lopen extra risico op pesterij. Moeite om de aandacht te houden bij doen en laten, al of niet in combinatie met druk gedrag (zoals bij ADHD) kan bijdragen tot onvoldoende functioneren in de omgang met anderen.
In vergelijking met gezonde leeftijdgenoten beoordelen kinderen met NF1 op schoolleeftijd hun aan gezondheid gerelateerde kwaliteit van leven als lager (Jennekens-Schinkel & Jennekens, 2008, pp 504-506).
fajenn31 mei, 2011 - 21:36
Begeleiding
Begeleiding
Vrijwel alle kinderen met NF1 behoeven de ene of andere vorm van extra bijstand om tot optimale ontplooiing te komen. Van belang is dat men niet voetstoots uitgaat van mindere intelligentie, want de helft van de kinderen met NF1 heeft een normaal IQ. De aard van de benodigde hulp kan met de leeftijd veranderen. Ze is ook niet voor elk kind met NF1 dezelfde.
Ouders en school kunnen geholpen worden om ongewenst gedrag, tegenvallende leervorderingen, emotionele reacties van het kind met NFG1 te begrijpen. Onderzoek door kinder-, neuro- of ontwikkelingspsychologen met kennis over en ervaring met NF1 kan een uitgangspunt opleveren voor aanpassingen in de leefwereld of voor therapie.
Meestal is het ongewenste gedrag niet alleen een gevolg van de ziekte maar zeker ook van omgang met het kind en van te hoge of te lage eisen die men aan het kind stelt. In dat geval kan de omgeving leren zich anders op te stellen. Slaaptekort of overmatig druk gedrag en/of aandachtsstoornis kunnen medicamenteus behandeld worden, zodat ze niet ook nog eens gevolgen hebben voor het leren (zie volgende punt). Eén van de zes kinderen met NF1 ontvangt in de schoolleeftijd medicatie vanwege ADHD.
Leerproblemen kunnen van algemene aard zijn, maar soms zijn ze beperkt tot bepaalde leervakken. Bij duidelijkheid over aard en ernst van de leerproblemen kan besloten worden tot extra onderwijshulp of soms tot verandering van schooltype. Belangrijk is dat het kind ervaart wat succes is, met andere woorden dat het gesteld wordt voor taken die het met oefening aankan! Ruim 80% van de NF1 kinderen in het reguliere basisonderwijs ontvangt extra onderwijsondersteuning. Als de leerproblemen ontstaan door onvoldoende slaap of aandachtstekort dan moet die oorzaak worden aangepakt.
Emotionele reacties kunnen te maken hebben met (bepaalde symptomen van) NF1, met de beperkingen die de ziekte meebrengt, maar ook met de ruimere levenssituatie van het kind. Voor de kleuter die naast een huisje een groot “schietding” tekende was niet de NF1 de belastende factor, ook al waren de verschijnselen aan het gelaat zichtbaar. Moeder was overspannen en kon de gezinstaken niet aan. Behandeling betekent in dit geval helpen de gezinssituatie te optimaliseren.
Op schoolleeftijd ontvangen grote minderheden van de Nederlandse kinderen met NF1 fysiotherapie vanwege motorische tekorten en logopedie vanwege spraaktekorten. Onderzoeken die aantonen dat motoriek en spraak daardoor verbeteren zijn nog niet beschikbaar (Krab et al., 2009).
Veel praktische adviezen kan men vinden in de brochure Leer- en Gedragsstoornissen van de hand van Dr LC Krab en anderen, zie www.neurofibromatose.nl.
fajenn31 mei, 2011 - 21:39
Een kind met neurofibromatose 1
Een kind met neurofibromatose 1
Veerle heeft een groot hoofd en zij ontwikkelt motorisch wat traag. Niet-familiaire NF1 wordt gediagnosticeerd als zij een jaar oud is; zij heeft dan CAL-vlekken en een klein gezwel op het rechter onderbeen. MRI van de hersenen toont als zij bijna vier jaar is gliomen in de hersenstam en in de kruising (chiasma) van de twee oogzenuwen. Niet te harden jeuk ontstaat in de rechter oorschelp, verspreidt zich over de rechterhelft van het gezicht en daarna over het hele lichaam. Een anti-jeuk medicijn helpt. Er ontstaat verlamming van de rechter stemband. Veerle krijgt vanaf het 9e jaar bijvoeding via een PEG-katheter. Het opticusglioom wordt groter. Veerle verliest het gezichtsvermogen bovenin het rechtergezichtsveld. Zij struikelt vaak. Slijm en benauwdheid geven haar vooral als zij verkouden is het gevoel te stikken. ‘s Nachts wordt zij bang om dood te gaan.
Veerle ontvangt spraaklessen en vanwege de nachtelijke angst speltherapie. Zij krijgt een gemotoriseerde driewieler. Aanvankelijk volgt zij basisonderwijs zonder moeite maar na enige tijd gaat zij over naar een school voor Langdurig Zieke Kinderen (LZK). In haar 10de jaar beschrijven haar ouders haar als wilskrachtig, ordelijk en rustig. Maar zij heeft geen vriendinnetjes meer. Zij reageert op afwijzing door leeftijdgenootjes met verdriet en hevige teleurstelling.
Zij wordt voor het eerst neuropsychologisch onderzocht als zij 4 jaar oud is. Zij functioneert dan ruim op leeftijdsniveau, maar natekenen van geometrische figuren lukt onvoldoende en mondt uit in gekras. Bij heronderzoek op de leeftijd van 6 jaar heeft zij een Verbaal IQ van 90 en een Performaal IQ van 82.
In haar 10de jaar is zij ongewoon mager en heeft zij zichtbare CAL-vlekken onder andere in de halsstreek. Het onderzoek vermoeit haar sterk. Het Verbale IQ bedraagt nu 85 en het Performale 75 (Wechsler Intelligence Scale for Children-Revised). De scores op de subtests zijn zeer wisselvallig. Een codeertaak bijvoorbeeld voert zij foutloos maar zo langzaam uit dat de score laag is. Haar reactietijden zijn lang. Bij leren van locaties op een computerscherm begint zij slecht. Na een interval blijkt zij het geleerde goed te hebben onthouden. Ook een gelezen tekst vertelt zij goed na. Zij past in een zinnendictee de spellingsregels goed toe maar zij is zwak in rekenen. Haar handschrift is fors en onbehouwen. Handvastheid en handvaardigheid (Motorische Prestatiebatterij) zijn onvoldoende. Zij tekent en kopieert geometrische figuren onder leeftijdsniveau.
Zij is 12 jaar als een glioom uit de pons wordt verwijderd. De jeuk verdwijnt, sondevoeding kan worden gestaakt. Zij volgt voortgezet middelbaar beroepsonderwijs in een school van cluster 3.
Figuur 6. Dictee in het 10de jaar.Figuur 7. De figuren van de Bender Visual Motor Gestalt test zoals deze door Veerle in haar 10de jaar zijn nagetekend op een vel blank A4 papierFiguur 8. De figuren van de Bender Visual Motor Gestalt zoals deze achtereenvolgens ter natekening aan Veerle zijn aangeboden
fajenn31 mei, 2011 - 21:41
Literatuur
Literatuur
Alivuotila L, Hakokari J, Visnapuu V, Korpijaakko-Huuhka A-M, Aaltonen O, Happonen R-P, Peltonen S, Peltonen J (2010) Speech characteristics in neurofibromatosis type 1. American Journal of Medical Genetics Part A 152A: 42-51
Boyd KP, Korf BR, Theos A (2009) Neurofibromatosis type 1. Journal of the American Academy of Dermatology 61: 1-16
DeBella K, Szudek J, Friedman JM (2000) Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 105: 608-614
Descheemaeker M-J, Ghesquière P, Symons H, Fryns JP, Legius E (2005) Behavioural, academic and neuropsychological profile of normally gifted neurofibromatosis type 1 children. Journal of Intellectual Disability Research 49 part 1, 33-46
Evans DG (2009) Neurofibromatosis type 2: a clinical and molecular review. Orphanet Journal of Rare Diseases. 19: 4-16
Evers A, Resing W (2007) Het drijfzand van didactische leeftijdsequivalenten. De Psycholoog 42: 466-472
Ferner RE, Huson SM, Thomas N, Moss C, Wilshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A (2007) Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. Review. Journal of Medical Genetics 44: 81-88
Gilboa Y, Josman N, Fattal-Valevski A, Toledano-Alhadef, Rosenblum S (2010) The handwriting performance of children with NF1. Research in Developmental Disabilities 31: 929-935
Huson SM, Korf BR (2002) The phakomatoses. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, Emery ALH (2002) Emery and Rimoin’s Principles & Practice of Medical Genetics 4th edition, volume 3, NewYork: Churchill Livingstone pp 3162-3202
Hyman SL, Shores A, North KN (2005) The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology 65: 1037-1044
Jennekens-Schinkel A, Jennekens FGI (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam: Uitgeverij Boom, Hoofdstuk 12, pp 487-512
Johnson H, Wiggs L, Stores G, Huson SM (2005) Psychological disturbance and sleep disorders in children with neurofibromatosis type 1. Developmental Medicine &Child Neurology 47: 237-242
Krab LC, Aarsen FK, de Goede-Bolder A, Catsman-Berrevoets CE, Arts WF, Moll HA, Elgersma Y (2008) Impact of neurofibromatosis type 1 on school performance. Journal of Child Neurology 23: 1002-1010
Krab, LC, de Goede-Bolder A, Aarsen FK, Catsman-Berrevoets CE (2009) Leer- en gedragsstoornissen bij kinderen met neurofibromatose type 1. Brochure gemaakt in opdracht van de Neurofibromatose Vereniging Nederland.
Krab LC, de Goede-Bolder A, Aarsen FK, Moll HA, de Zeeuw CI, Elgersma Y, van der Geest JN (2011) Motor learning in children with neurofibromatosis type 1. Cerebellum 10: 14-21
Lammert M, Friedman JM, Kluwer L, Mautner VF (2005) Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Archives of Dermatology 141: 78-79
McKeever K, Shepherd CW, Crawford H, Morrison PJ (2008) An epidemiological, clinical and genetic survey of neurofibromatosis type 1 in children under sixteen years of age. Ulster Medical Journal 77: 160-163
Pasmant E, Masliah-Planchon J, Lévy P, Laurendeau I, Ortonne N, Parfait B, Valeyrie-Allanore L, Leroy K, Wolkenstein P, Vidaud M, Vidaud D, Bièche I (2011) Identification of genes potentially involved in the increased risk of malignancy in NF1-microdeleted patients. Molecular Medicine 17: 79-87
Payne JM, Hyman SL, Shores EA, North KN (2011) Assessment of executive function and attention in children with neurofibromatosis type 1: relationships between cognitive measures and real-world behavior. Child Neuropsychology 1:1-17
Payne JM, Moharir MD, Webster R, North KN (2010) Brain structure and function in neurofibromatosis type 1: current concepts and future directions. Journal of Neurology, Neurosurgery and Psychiatry 81: 304-309
Sabbagh A, Pasmant E, Laurendeau I, Parfait B, Barbarot S, Guillot B, Combemale P, Ferkal S, Videaud M, Aubourg P, Vidaud D, Wolkenstein P and members of the NF France network (2009) Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Human Molecular Genetics 18: 2768-2778
Thompson HL, Viskochil DH, Stevenson DA, Chapman KL (2010) Speech−language characteristics of children with neurofibromatosis type 1. American Journal of Medical Genetics Part A 152A: 284-290
Van Engelen SJ, Krab LC, Moll HA, de Goede-Bolder A, Pluijm SM, Catsman-Berrevoets CE, Elgersma Y, Lequin MH (2008) Quantitative differentiation between healthy and disordered brain matter in patients with neurofibromatosis type 1 using diffusion tensor imaging. American Journal of Neuroradiology 29: 816-822
Tubereuze sclerose complex (TSC) behoort tot de neurocutane aandoeningen. De ziekte treft de hersenen en de huid; maar ook nieren, longen en andere organen kunnen bij de ziekte betrokken zijn. Een “tuber” is een gezwel of bult, “sclerose” betekent verharding en “complex” is aan de naam toegevoegd vanwege de grote verscheidenheid aan verschijnselen van de ziekte. De hersenaandoening uit zich vooral in epilepsie, zwakzinnigheid en autisme. Toch worden deze verschijnselen niet gerekend tot de diagnostische kenmerken omdat ze bij zeer veel hersenaandoeningen voorkomen en dus niet bijdragen tot het onderscheid van TSC met andere ziekten.
In deze tekst wordt eerst vermeld op welke andere websites men voor deskundige informatie over TSC terecht kan. Vervolgens worden de lichamelijke verschijnselen en de medische behandeling kort samengevat. Daarna worden cognitie en gedrag, psychosociale problemen en begeleiding van kinderen met TSC besproken.
fajenn7 juli, 2011 - 12:58
Andere teksten op internet
Andere teksten op internet
www.kinderneurologie.eu: de Nederlandstalige tekst is geschreven door een kinderneuroloog. De informatie is vooral bestemd voor ouders en anderszins geïnteresseerden. www.tuberous-sclerosis.org: de site van de Britse patiëntenvereniging omvat een deel voor zorgverleners die beroepshalve bij TSC zijn betrokken (“professionals”). Via dat deel kan men toegang krijgen tot “Clinical guidelines for the care of patients with tuberous sclerosis complex” en tot de “Assessments of Behaviour and Learning in Tuberous sclerosis complex”. De klinische zorgrichtlijnen zijn in 1998 opgesteld en sedertdien incidenteel geactualiseerd. De tekst over onderzoek van gedrag en leren dateert van een conferentie in 2003 en is in 2005 gepubliceerd.
fajenn7 juli, 2011 - 13:06
Epidemiologie
Epidemiologie
Algemeen wordt opgegeven dat TSC voorkomt bij ongeveer 1 per 6000 levend geborenen (geboorteincidentie). Gegevens van recent onderzoek ontbreken.
fajenn7 juli, 2011 - 13:08
Erfelijkheid en genmutaties
Erfelijkheid en genmutaties
TSC erft autosomaal dominant over. De penetrantie is 100%. In bijna zeven van de tien gevallen is de ziekte echter “geïsoleerd”, dat wil zeggen niet aanwezig bij vader of moeder. Bij deze geïsoleerde gevallen is een mutatie ontstaan in het DNA van cellen in de kiemcellijn: in de onbevruchte eicel, de onbevruchte zaadcel of de bevruchte eicel in het “eencellige stadium”. Een aanmerkelijk percentage van de patiënten heeft de ziekte in zo ernstige mate dat zij geen nakomelingen verwekken (Orlova & Crino, 2010; Curatolo et al., 2008).
Het ene gen (het TSC1 gen) ligt op de lange arm van chromosoom 9 en het andere gen (het TSC2 gen) ligt op de korte arm van chromosoom 16. Bij geïsoleerde gevallen worden vaker TSC2 genmutaties dan TSC1 genmutaties gevonden. TSC2 mutaties hebben vaker ernstige effecten dan TSC1 mutaties. De mutaties in het TSC2 gen hebben soms het karakter van deleties waarbij ook een naastgelegen gen betrokken kan zijn. In die gevallen behoren cystevorming in de nieren en achteruitgang van de nierfunctie tot het ziektebeeld.
Het TSC1 gen bevat de code voor het eiwit hamartine en het TSC2 gen bevat de code voor het eiwit tuberine.
Hamartine en tuberine binden aan elkaar en vormen een heterodimeer; dit dimeer vermindert de stimulering van een ander eiwit dat cruciale functies heeft in tal van celprocessen. Dit eiwit staat bekend onder twee namen. “mTOR” (mammalian target of rapamycin) is de meest gebruikte naam. De tweede naam is “FRAP1” (FK506 binding protein 12-rapamycin-associated protein 1). Rapamycin is bekend als geneesmiddel in de transplantatiegeneeskunde. mTOR heeft een regulerende functie voor celproliferatie, celmotiliteit, celoverleving, en eiwitsynthese. mTOR integreert de werking van groeifactoren en het registreert voedings-, energie- en redox-toestand.
fajenn7 juli, 2011 - 13:12
Afwijkingen in organen
Afwijkingen in organen
Hersenen
In de hersenen komen drie soorten afwijkingen alleen of in combinaties voor (Curatolo et al., 2008) :
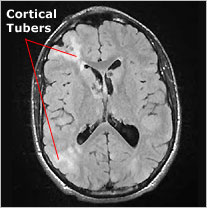
Harde tubers in de cortex en noduli (knobbeltjes) onder de bedekkende cellaag (ependym) van de hersenkamers (zijventrikels). in de tubers bevinden zich abnormale zenuwcellen, gliacellen en reuscellen. De noduli zijn soms verkalkt en daardoor makkelijk herkenbaar.
Figuur 1a. MRI, T1 gewogen. Afwijkingen zoals voorkomend bij tubers
Overgenomen via Google afbeeldingen van www.massgeneral.orgFiguur 1b. CT scan. Verkalkte noduli, subependymaal
Overgenomen via Google afbeeldingen van www.emedicine.medscape.com
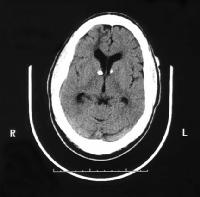
Subependymale reusceltumoren : knobbels nabij het foramen van Monro die neigen tot groter worden, tot afsluiten van het foramen van Monro en die daardoor een levensbedreigende toestand kunnen veroorzaken. Ze komen voor bij ongeveer 10% van de kinderen.
Figuur 2. MRI, T1 gewogen. Afwijking zoals voorkomend bij subependymale reuscel tumor
Overgenomen via Google afbeeldingen van www.neurol.org
Afwijkingen van de witte stof; onderzoek met een MRI techniek (“diffusion tensor imaging”, “DTI”) heeft aanwijzingen opgeleverd voor afwijkingen van met name het myeline (Krishnan et al., 2010).(Zie ENCYCL-Beeldvorming) Hoe die afwijkingen er onder de microscoop uitzien is nog niet gerapporteerd.
Andere organen
De afwijkingen in andere organen zijn niet minder complex dan die in de hersenen. Het meest komen hamartomen voor, goedaardige gezwellen met verschillende typen cellen met abnormale ordening van de cellen.
fajenn7 juli, 2011 - 13:15
Verschijnselen van belang voor de diagnose
Verschijnselen van belang voor de diagnose
Voor het stellen van de diagnose maakt men onderscheid tussen majeure (belangrijke) en mineure (mindere) verschijnselen (Curatolo et al., 2008; zie Jennekens-Schinkel en Jennekens, 2008). Men acht de diagnose zeker als het kind twee majeure verschijnselen of één majeur verschijnsel en twee mineure verschijnselen heeft. Genmutaties kunnen in 10 tot 15% van de kinderen niet worden aangetoond; mede daarom wordt DNA onderzoek niet altijd gedaan.
Majeure verschijnselen
ongepigmenteerde (“hypomelanotische”) huidvlekken; nemen toe in de kinderjarenFiguur 3. Hypomelanotische vlek:
Overgenomen via Google afbeeldingen van www.tsiv.be
in het gelaat rozerode tot blauwige knobbeltjes die bestaan uit bindweefsel en kleine vaatjes (angiofibromen) en ontstaan in de kinderjaren. Op het voorhoofd geelachtige, iets verheven plekken (“plaques”)Figuur 4. Angiofibromen:
Overgenomen via Google afbeeldingen van www.stsn.nl
onder de nagelrand of in de nagelgroeve bindweefselknobbeltjes (unguale of periunguale fibromen), vanaf ongeveer het 17de jaar
op de romp, vooral aan de rugzijde, segrijn plaques (plekken als van korrelig leer, met een geelbruin of roze aspect ter grootte van millimeters tot wel 10 cm), vanaf 10de jaar
op het netvlies platte of knobbelige gezwelletjes (hamartomen) zonder veel nadelige gevolgen voor het zien, ze ontstaan op kinderleeftijd
in de hersenen corticale tubers, subependymale noduli en subependymale reusceltumoren
in het hart rhabdomyomen (spiergezwellen die ontstaan tijdens de foetale periode en na de geboorte afnemen in aantal en grootte)
in de nieren verschillende soorten tumoren die neigen tot bloeden en daardoor levensgevaarlijk zijn. Cystenieren bij TSC2 gendeleties (Zie "Erfelijkheid en genmutaties")
Mineure verschijnselen
Putjes in het enamel van het gebit, fibromen in het tandvlees, poliepen in het rectum, cysten in het bot, kleurloze plekjes in het netvlies en andere verschijnselen.
fajenn8 juli, 2011 - 12:16
Neurologische en psychiatrische verschijnselen
Neurologische en psychiatrische verschijnselen
De overgrote meerderheid van de kinderen heeft stoornissen die rechtstreeks gevolg zijn van de hersenafwijkingen (Curatolo et al., 2008; zie Jennekens-Schinkel & Jennekens, 2008):
Epilepsie
Epilepsie komt voor bij bijna alle patiënten met TSC (80-90%). De aanvallen beginnen bij 60% van de kinderen al in het eerste levensjaar, veelal met partiële aanvallen en bij ongeveer 40% met “infantiele spasmen” (syndroom van West) (zie zowel voor partiële aanvallen als voor het syndroom van West Ziektebeschrijving Epilepsie). Deze spasmen verdwijnen na verloop van tijd maar enigerlei vorm van epilepsie blijft bestaan. De aanvallen kunnen als status epilepticus voorkomen. De aanvallen reageren bij twee van de drie kinderen niet of onvoldoende op behandeling met anti-epileptica (“refractaire epilepsie”) (Chu-Shore et al., 2010).
Zwakzinnigheid Zwakzinnigheid of verstandelijke beperking doet zich voor bij ongeveer 40% van de kinderen en is niet zelden zo ernstig dat psychologisch testonderzoek met de gebruikelijke instrumenten niet mogelijk is. Zie verder de paragraaf over "Cognitief functioneren".
Stoornissen behorend tot het “autismespectrum”
Stoornissen behorende tot het autismespectrum zijn in het algemeen gekenmerkt door
onvoldoende en gestoorde sociale interactie en communicatie
herhalingsgedrag
smal interessegebied
In de bevolking komt autisme voor bij 1% van alle kinderen, en wel vooral bij kinderen met uiteenlopende genetische en niet genetische aandoeningen die gepaard gaan met zwakzinnigheid (Moss & Howlin, 2009). Zeer veel genen kunnen betrokken zijn bij stoornissen in het autismespectrum. Voor de diagnose “autisme” of “stoornis in het autismespectrum” is geen enkelvoudig hard criterium beschikbaar. De diagnose wordt tegenwoordig meestal gesteld met behulp van vragenlijsten (Moss & Howlin, 2009; zie ook Jennekens-Schinkel & Jennekens, 2008, pp. 525-526).
Twintig tot zestig procent van de kinderen met TSC heeft verschijnselen van autisme en stoornissen in het autismespectrum. Waardoor de opgegeven percentages zo variëren is niet met zekerheid bekend, aannemelijk is dat de diagnostiek tekort schiet en/of dat de samenstelling van de onderzochte groepen verschilt. Niet zelden heeft een kind zowel epilepsie, stoornissen in het autismespectrum als zwakzinnigheid.
Agressiviteit, angst, depressie
Agressiviteit is een lastig woord. Het kan verwijzen naar “neiging tot geweld”, maar ook naar “destructief gedrag”, “opwinding of onrust” of “vijandig gedrag of dreigende houding met de bedoeling een dominante positie af te dwingen of het eigen terrein te verdedigen”. Angst kan leiden tot agressieve reacties, maar ook tot vluchtreacties. Agressiviteit, angst en depressiviteit zijn bij kinderen met TSC gerapporteerd. Weinig gegevens zijn beschikbaar over welke aspecten van de genoemde begrippen in welke mate en bij welke kinderen voorkomen. Ook verband met andere neurologische en gedragsafwijkingen zijn onvoldoende bekend.
Auto-mutilatie (“self-injury”) kan een probleem vormen bij zwakzinnige, autistische kinderen met TSC (Staley et al., 2008).
Slaapproblemen
Slaapproblemen worden vooral waargenomen bij kinderen met nachtelijke aanvallen van epilepsie. Niet duidelijk is of ze een stoornis zijn, rechtstreeks samenhangen met de epilepsie, of secundair (bijvoorbeeld door vrees om een nachtelijke aanval te krijgen uit de slaap gehouden worden). In het laatste geval zijn heel andere maatregelen geboden dan in het eerste geval.
fajenn7 juli, 2011 - 13:30
Cognitief functioneren
Cognitief functioneren
Mentale ontwikkeling en intelligentie
Ontwikkeling van zuigelingen en peuters toont grote verschillen en is bij veel kinderen vertraagd (zie voor overzicht Jennekens-Schinkel & Jennekens, 2008, pp 522–5). Het ontwikkelingsquotiënt varieert bij kinderen in deze leeftijdsperiode van minder dan twintig tot meer dan 100 (het gemiddelde in de kinderbevolking is 100, met een spreiding tussen 85 en 115).
Intelligentie. Als de kinderen op een leeftijd zijn gekomen dat de intelligentie met de daarvoor gangbare tests onderzoekbaar is, blijkt in verschillende groepen kinderen met TSC dat de verdeling van het Totale IQ (TIQ niet zoals normaal één top heeft, maar twee toppen: een top bij ongeveer 92 en een top in het gebied van ongeveer 45 tot 20 of minder. Ruim 50% van de kinderen met TSC heeft een Totaal IQ boven 70, oplopend tot 130
Gezonde broertjes en zusjes, ook van kinderen met TSC die een IQ in het normale gebied hebben, functioneren in de regel op een hoger intelligentieniveau
Andere cognitieve domeinen Specifieke stoornissen zijn niet met zekerheid aangetoond. Zwak functioneren in domeinen als geheugen of taal passen meestal bij het niveau van de intelligentie. Aandacht is misschien een uitzondering. Kinderen met TSC hebben meer moeite om hun aandacht bij spel of werk te bepalen dan gezonde broertjes/zusjes. In testonderzoek blijkt dit zowel in “selectief aandacht geven”, “volhouden van aandacht”, “wisselen van instelling”, en “onderdrukken van niet meer juiste reacties”. Dubbeltaken zijn het moeilijkst (de Vries et al., 2009). Aandachtsprobleem al of niet met hyperactiviteit (attention deficit hyperactivity disorder) Afleidbaarheid en onoplettendheid in combinatie met ongewoon druk en impulsief gedrag worden samengevat als “attention deficit hyperactivity disorder” (ADHD) als de combinatie zich in meer dan één situatie voordoet, dus niet alleen thuis maar ook bijvoorbeeld op school en/of bij sport- of andere vrijetijdsactiviteiten. Een eenvoudig hard criterium voor ADHD ontbreekt. Voor de diagnostiek wordt gebruik gemaakt van gestandaardiseerde vragenlijsten. Vijftig procent of meer van de kinderen met TSC, in het bijzonder de zwakzinnige kinderen, heeft gedrag dat past bij ADHD (Jennekens-Schinkel & Jennekens, 2008, pp 525-6). Waardoor zoveel ernstige stoornissen van cognitief functioneren Een geheel bevredigende verklaring is er nog niet. Als er ernstige stoornissen van het cognitieve functioneren zijn, dan worden deze tot nu toe begrepen uit een drietal factoren:
Genmutaties: de stoornissen van de cognitie tenderen naar grotere ernst bij TSC2- dan bij TSC1-genmutaties
Hersenafwijkingen: het aantal cerebrale tubers heeft geen sterk verband met de intelligentie. Wel lijkt er enig verband te zijn tussen het door tubers in beslag genomen hersenvolume en de epilepsie, maar dit verband verklaart op zichzelf de ernst van de cognitiestoornissen niet. Afwijkingen rondom de tubers zouden ook van belang kunnen zijn
Infantiele spasmen en epilepsie: Kinderen met infantiele spasmen en medicamenteus onvoldoende behandelbare (refractaire) epilepsie hebben meestal een laag IQ. Toch zijn er kinderen met infantiele spasmen en vroeg begonnen epilepsie die een normale intelligentie hebben (Chu-Shore et al., 2010; de Vries et al., 2009)Figuur 5. Leeftijd eerste aanval en intelligentie bij 36 kinderen en 18 volwassenen: Overgenomen uit Jennekens-Schinkel en Jennekens, 2008, pg 528
Anti-epileptische medicatie en slecht slapen kunnen mede een rol spelen, maar geen van beide factoren verklaart de ernst van de cognitiestoornissen
fajenn7 juli, 2011 - 13:36
Schoolloopbaan
Schoolloopbaan
Over de schoolloopbaan van niet-zwakzinnige TSC kinderen zijn weinig gegevens beschikbaar.
fajenn7 juli, 2011 - 13:38
Ziektebeloop, medische controle en behandeling
Ziektebeloop, medische controle en behandeling
De ernst van de ziekte varieert sterk. Veel verschijnselen verergeren in de loop der jaren. Bij geboorte hebben sommige kinderen tot regressie neigende hartspiergezwellen, en hartritmestoornissen. Hypomelanotische plekken zijn veelal vanaf de eerste levensjaren aanwezig. Later ontstaan de andere huidafwijkingen. In het eerste jaar verschijnen bij de meerderheid van de kinderen epileptische aanvallen.(Zie Ziektebeschrijving epilepsie). Tachtig procent van de kinderen van 3 jaar en ouder heeft epilepsie. Tot bloeding neigende niertumoren doen zich meestal voor in de puberteit of later. Longafwijkingen ontstaan pas op volwassen leeftijd.
De medische zorg wordt veelal gecoõrdineert door een kinderneuroloog. Aan de behandeling nemen verschillende specialsten deel. Zorg en behandeling richten zich op de epilepsie, de psychiatrische stoornissen, de huidafwijkingen, de hartritmestoornissen en vooral op de mogelijk levensbedreigende complicaties, zoals bloedingen uit vaatrijke gezwellen in de nieren en verhoogde intracraniële druk door groei van subependymale reuscel tumoren die hydrocefalus kunnen veroorzaken. De medicamenteuze behandeling van epilepsie heeft bij de meerderheid van de kinderen onvoldoende succes. Voor refractaire epilepsie kan epilepsiechirurgie overwogen worden. Angiofibromen in het gelaat kunnen behandeld worden met laser- of cryochirurgie. Advies betreffende slaapproblemen (zie verderop) is van belang voor het gedrag overdag.
fajenn7 juli, 2011 - 13:40
Begeleiding
Begeleiding
Bij ongeveer 50% van de kinderen met TSC doen zich geen grote problemen voor
TSC kan echter gedurende lange tijd tot een reeks van problemen aanleiding geven. Begeleiders kunnen ouders bijstaan met praktische adviezen aangaande leefwijze, spel, stimulering van de cognitie en opvoeding tot eigen verantwoordelijkheid van het kind.
Alle gedragsproblemen die zich voordoen bij slaapstoornissen, epilepsie, autisme, ADHD en zwakzinnigheid kunnen afzonderlijk of in combinatie voorkomen bij kinderen met TSC en behoren op de daarbij passende wijze te worden aangepakt. Medicatie of verbetering van medicatie valt te overwegen bij slaapstoornissen, ADHD en epilepsie
TSC kan bij uitzondering aanleiding geven tot zeer complexe en hardnekkige gedragsproblemen waar oplossingen moeilijk voor te bedenken zijn. In zulk geval kunnen zorgverleners via het Centrum voor Consultatie en Expertise (www.cce.nl ) een beroep doen op het TSC-Expertiseteam
De huidafwijkingen kunnen leiden tot gevoelens van schaamte en tot belemmering van het sociale functioneren. Alleen al om deze reden moeten begeleiders aandringen op vroegtijdige dermatologische behandeling
Voor advies over onderwijs en schoolkeuze is deskundige evaluatie van het cognitieve en sociale functioneren nodig wanneer de ontwikkeling van het kind met TSC daar aanleiding toe geeft. Te beantwoorden vragen zijn onder andere: wat kan het kind cognitief aan, hoe kan het cognitieve functioneren het beste worden gestimuleerd, hoe kan het kind tot succes worden gebracht in sport en spel. Herbeoordeling is gewenst als zich een probleem voordoet in de schoolcarrière
fajenn7 juli, 2011 - 13:43
Een adolescent met tubereuze sclerose
Een adolescent met tubereuze sclerose
Jens heeft al bij zijn geboorte niet-gepigmenteerde vlekken op de huid. De eerste epileptische aanval doet zich voor in zijn zesde levensjaar. Onderzoek leidt tot de diagnose TSC als gevolg van een mutatie in het TSC1 gen. Verschillende anti-epileptica worden geprobeerd, maar aanvallen blijven voorkomen. Cognitief en sociaal functioneert Jens goed. Zijn schoolloopbaan verloopt naar wens.
In de 6de klas van het gymnasium heeft hij twee tot drie aanvallen per week. Hij voelt een aanval aankomen doordat hij de toon van geluiden als verhoogd waarneemt. Bij doorzetten van de aanval hoort hij een stem die onaangename opdrachten geeft of mededelingen doet, hij voelt zich als in een draaikolk, zijn bewustzijn daalt, hij krijgt een globale tonische kramp, de pupillen worden wijd, hij wordt angstig en hij roept om hulp. Na drie minuten is de aanval voorbij. Hij is dan moe, maar hij hervat meestal zijn bezigheden.
Jens meldt zich aan voor epilepsiechirurgie. MRI (ENCYCL-beeldvorming toont verspreid in de hersenen verschillende tubers die passen bij TSC (zie Figuur 1 in de paragraaf "Afwijkingen-in-organen"). In het basale deel van de rechter temporaalkwab wordt bij EEG een epileptisch focus gezien. Neuropsychologisch onderzoek voorafgaande aan operatie toont een Totaal IQ (TIQ van 119, Verbaal IQ 117, Performaal IQ 118. Aanwijzingen voor globale of specifieke functiestoornissen ontbreken. Verwijdering van het rechts temporale gebied levert blijkens de Wada-test geen risico op voor geheugen en taal.
Postoperatief doen zich geen aanvallen meer voor. De taalfunctie blijft onveranderd. Enige daling in de globale index van geheugen en leren is 24 maanden na epilepsiechirurgie weer geheel verdwenen. Jens verkeert dan in de studentenwereld waar hij zich goed aanpast. Hij presteert naar behoren in zijn studie, bespeelt een muziekinstrument en doet aan sport en spel. Hij leeft meer ontspannen dan voorheen.
fajenn7 juli, 2011 - 13:45
Literatuur
Literatuur
Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA (2010) The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 51: 1236-1241
Jennekens-Schinkel A, Jennekens FGI (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam, Uitgeverij Boom
Krishnan ML, Commowick O, Jeste SS, Weisenfeld N, Hans A, Gregas MC, Sahin M, Warfield SK (2010) Diffusion features of white matter in tuberous sclerosis with tractography. Pediatric Neurology 42: 101-106
Moss J, Howlin P (2009) Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. Journal of Intellectual Disability Research 53: 852-873
Orlova KA, Crino PB (2010) The tuberous sclerosis complex. Annals of the New York Academy of Sciences 1184: 87-105
Staley BA, Montenegro MA, Major P, Muzykewicz DA, Halpern EF, Kopp CMC, Newberry P, Thiele EA (2008) Self-injurious behavior and tuberous sclerosis complex: frequency and possible associations in a population of 257 patients. Epilepsy & Behavior 13: 650-653
de Vries PJ, Gardiner J, Bolton PF (2009) Neuropsychological attention deficits in tuberous sclerosis complex (TSC). American Journal of Medical Genetics Part A 149A: 387-395