Metachromatische leukodystrofie
Metachromatische leukodystrofie fajenn 6 februari, 2011 - 15:35Inleiding
InleidingMetachromatische leukodystrofie (MLD) is een stapelingsziekte, een van de vele zeldzame erfelijke stoornissen van de stofwisseling. Door tekort aan activiteit van een enzym hoopt zich een afbraakproduct in het weefsel op. Bij kleuring van zenuwweefsel voor microscopisch onderzoek (zoals vroeger gebruikelijk voor diagnostiek) blijkt het afbraakproduct een andere kleur te hebben dan de omgevende structuren. Dat verklaart de term metachromatisch (anderskleurig). Men spreekt van leukodystrofie omdat het vooral een aandoening is van de witte stof in het zenuwweefsel. Van lang niet alle erfelijke stofwisselingsziekten zijn neuropsychologische gegevens beschikbaar. In beperkte mate is dat wel het geval bij MLD.
Na enkele inleidende paragrafen zal eerst een korte samenvatting van de lichamelijke verschijnselen, van de huidige diagnostiek en van de therapie worden geboden waarna nader zal worden ingegaan op cognitieve en gedragsmatige aspecten. De tekst wordt afgesloten met een ziektegeschiedenis.
Andere teksten op internet
Andere teksten op internetwww.kinderneurologie.eu: biedt een uitvoerige Nederlandstalige beschrijving van het ziektebeeld door een kinderneuroloog.
www.ncbi.nlm.nih.gov/OMIM: De Engelstalige site over erfelijke ziekten van de John Hopkins Universiteit in de VS.
www.stofwisselingsziekten.nl: een fraai verzorgde Nederlandstalige site waarin ook MLD aan de orde komt.
Epidemiologie
EpidemiologieUit studies in de VS en Nederland blijkt dat de prevalentie bij geboorte tussen 1 en 1.5 per 100.000 bedraagt (Bonkowski et al., 2010; Poorthuis et al., 1999). In genetisch geïsoleerde groepen mensen worden veel hogere prevalenties aangetroffen.
Erfelijkheid en genmutatie
Erfelijkheid en genmutatieDe ziekte is autosomaal recessief erfelijk.
- In de grote meerderheid der gevallen wordt de ziekte veroorzaakt door een mutatie in het gen dat de code bevat van het enzym arylsulfatase A op chromosoom 22. Als beide ouders een gemuteerd arylsulfatase A gen dragen heeft ieder van hun kinderen een kans van 25% op MLD. Er zijn veel verschillende mutaties van het arylsulfatase A gen bekend.
- Het arylsulfatase A is nodig voor de afbraak van een sulfatide verbinding. De sulfatide verbinding moet gemobiliseerd worden, beschikbaar gemaakt, wil afbraak kunnen plaats vinden. Voor de mobilisatie is een activator (saponine B) nodig. Ontbreekt deze, dan kan MLD het resultaat zijn (Gieselmann en Krägeloh-Mann, 2010).
Oorzaak
OorzaakArylsulfatase A is een lysosomaal enzym. Ontbreekt arylsulfatase A of is er een deficiëntie of ontbreekt saponine B dan hoopt het niet afgebroken chemisch product op in de lysosomen van oligodendrogliacellen en Schwanncellen, en in enige mate in zenuwcellen. Oligodendroglia en Schwanncellen zijn betrokken bij de aanmaak van myeline in het centrale- respectievelijk perifere zenuwstelsel. Wordt er al teveel gestapeld dan volgt afbraak van het myeline, dat wil zeggen van witte stof. Hoe de stapeling myeline afbraak (demyelinisatie) tot gevolg kan hebben is niet bekend. De ziekteverschijnselen van MLD worden geheel of vrijwel geheel toegeschreven aan de demyelinisatie. De ernst hangt af van de mutaties. Soms ontbreekt activiteit van arylsulfatase A geheel, soms is er nog restactiviteit (Gieselmann-Krägeloh-Mann, 2010).
Lichamelijke verschijnselen
Lichamelijke verschijnselenEnigszins schematiserend worden drie vormen van de ziekte onderscheiden (Gieselmann & Krãgeloh-Mann, 2010; Jennekens-Schinkel & Jennekens, 2008).
Laat infantiele vorm
Bijna de helft van alle gevallen van MLD behoort tot deze vorm. De eerste verschijnselen treden meestal op in de loop van het tweede levensjaar, in ieder geval voor de leeftijd van 3 jaar (vanaf negende tot 27ste maand bij 21 kinderen; Kehrer et al., 2011). Het beeld begint met symptomen die passen bij een aandoening van het perifere zenuwstelsel: de kinderen leren wel lopen maar de spieren worden slap en verzwakken, het lopen gaat snel achteruit en de peesreflexen verdwijnen. Later worden de spieren stijf en atrofisch, er ontstaan pathologische reflexen, het kind kan niet meer zitten, de spraak wordt dysartrisch, het contact vermindert. Blindheid, aanvallen van epilepsie en strekkrampen (verschijnselen van motorische ontremming) komen voor. De kinderen overlijden vaak omstreeks het 6de levensjaar, maar het beloop kan ook korter of veel langer zijn.
Juveniele vorm
In 30-40% van de ziektegevallen past het beloop bij de juveniele vorm. De eerste verschijnselen ontstaan tussen het tweede tot derde en 16de jaar (bij 38 kinderen tussen 3 en 14 jaar; Kehrer et al., 2011). Symptomen van het perifere zenuwstelsel ontbreken of staan op de achtergrond. Leer- en gedragsstoornissen kunnen de beginverschijnselen zijn. Het ziektebeloop is langzamer dan bij de laat infantiele vorm; de ziekteduur kan wel twintig jaar bedragen.
Adulte vorm
De ziekte presenteert zich in het algemeen met gedragsveranderingen, cognitieve stoornissen, psychose of epilepsie. In de loop van tientallen van jaren verergert het beeld zeer geleidelijk.
Diagnose
DiagnoseDe demyelinisatie van perifere zenuwen wordt meestal ontdekt bij onderzoek van hun geleidingssnelheid. De aantasting van het myeline (witte stof) in de hersenen blijkt bij MRI. In de urine wordt de abnormale uitscheiding van sulfatiden aangetoond en in het bloed de enzymdeficiëntie. De genmutatie kan door DNA-onderzoek worden aangetoond. Kinderen bij wie de ziekte zich presenteert met veranderingen van cognitie en gedrag worden vaak pas na geruime tijd goed gediagnosticeerd. In de tussentijd kunnen tal van vergissingen worden gemaakt in interpretatie en behandeling van de gedragsveranderingen.
Medische behandeling en preventie
Medische behandeling en preventieVoor zover de ziekte kan worden tegengegaan, gebeurt dat door hematopoëtische stamcel transplantatie (zie ook ENCYCL-hematopoëtische stamcel transplantatie). Bij deze behandeling ontwikkelen cellen met het intacte gen afkomstig van het beenmerg zich in het centrale zenuwstelsel tot microglia. Geruime tijd verstrijkt voordat deze microglia effectief is. Eenmaal ontstane afwijkingen herstellen niet of onvoldoende, progressie kan worden tegengegaan. Voor de laat infantiele vorm kost dit therapieproces teveel tijd. Onderzoek over het therapeutisch nut van enzymtoediening of van gentherapie is gaande (Gieselmann & Krägeloh-Mann, 2010; Pierson et al., 2008).
Voor tal van symptomen (ziekteverschijnselen) is behandeling wel mogelijk, bijvoorbeeld voor pijn, spasmen, psychose, epilepsie.
Preventie van de ziekte is mogelijk door vruchtwateronderzoek in het begin van de zwangerschap en zo nodig abortus.
Cognitie en gedrag
Cognitie en gedragHet cognitieve verval verloopt met verschillende snelheid, niet alleen bij de diverse vormen van de ziekte maar ook bij vergelijking van twee aangedane kinderen met dezelfde genetische afwijking in een gezin. De organische basis van aandachtstoornissen en gedragsproblemen wordt in de aanvangsfase vaak langdurig miskend.
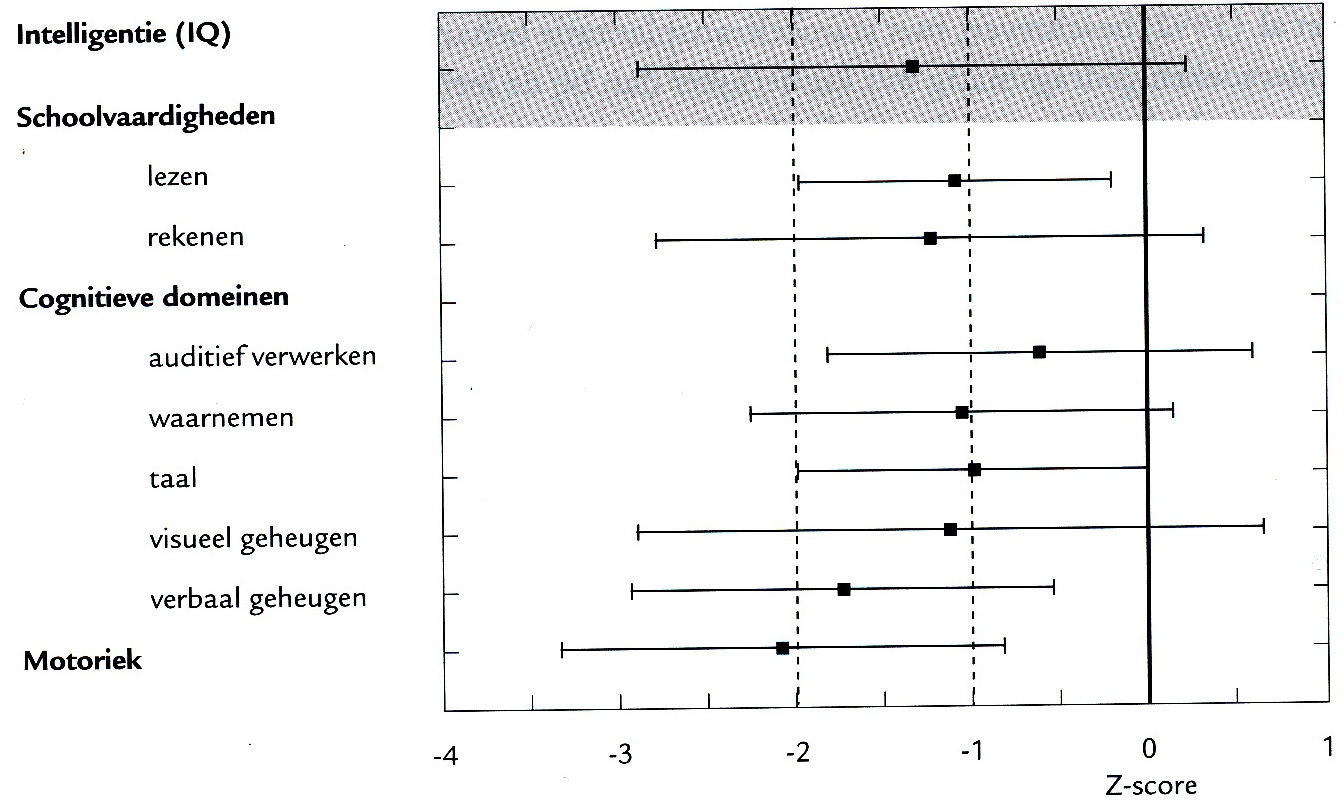
In onderstaande figuur zijn bevindingen weergegeven van cognitief en motorisch testonderzoek van 21 kinderen (leeftijd tussen 2 en 7 jaar) die verkeerden in de diagnostische fase van de ziekte. Allen hadden beginsymptomen voor de leeftijd van 6 jaar. De cognitieve domeinen (auditief verwerken, waarnemen, taal, verbaal geheugen en visueel geheugen) werden onderzocht met verschillende, maar leeftijdsadequate gestandaardiseerde tests. Voor onderlinge vergelijkbaarheid van de test uitslagen werden de scores getransformeerd in z-scores (gemiddeld = 0, standaarddeviatie = 1. Alle gemiddelden liggen onder het normgemiddelde, de spreiding is in alle domeinen fors, de motoriek is het meest gestoord. Beperkte woordenschat, overmatig zoeken naar woorden, gebruik van lege woorden en wijdlopigheid kenmerken het mondelinge taalgebruik. Echte afasie komt zelden of niet voor.
Begeleiding
Begeleiding- Diagnostische en therapeutische onzekerheid zijn moeilijk te verwerken, dat geven zowel ouders als zieke kinderen aan.
- Ouders en zusjes of broers behoeven bijstand en informatie over het te verwachten ziektebeloop - ook ten aanzien van cognitie en gedrag - en de moeilijkheden die zich daarbij kunnen voordoen.
- Het zieke kind dient zo lang en zoveel mogelijk zelfstandig te functioneren.
Een kind met metachromatische leukodystrofie
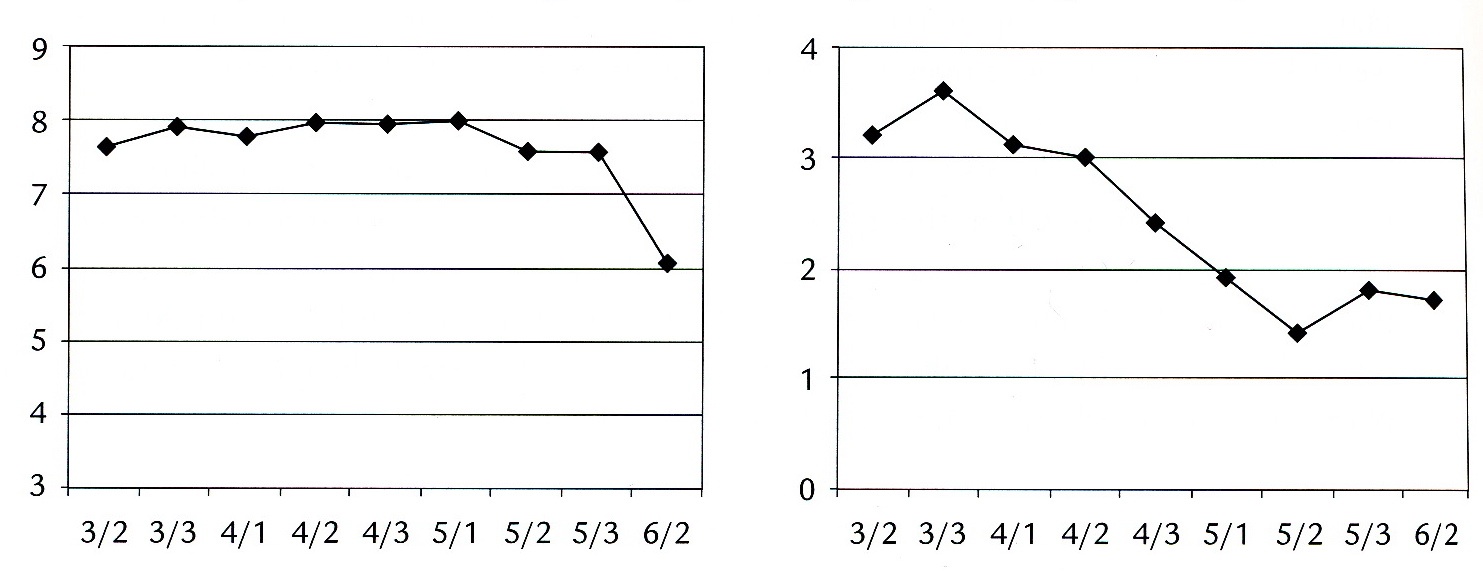
Een kind met metachromatische leukodystrofieJob was tot omstreeks zijn 9e jaar een opgewekte, sociaal ingestelde jongen. Op school was hij tot groep 6 een goede leerling. Hij veranderde echter. Hij kreeg conflicten, ging in bed plassen en presteerde steeds minder op school. (Figuur 2 toont beloop van leervorderingen en gedrag.) Voor dat alles was geen oorzaak aanwijsbaar. Zijn ouders merkten dat hij veel op zijn tenen liep en sleepte met een been. Aanvankelijk werden de diagnoses PDD-NOS en syndroom van Rett overwogen. MLD werd gediagnosticeerd toen hij 10 jaar was. Ouders en brusjes waren gezond; de ouders hadden een hogere opleiding genoten en werkten dienovereenkomstig.
Neuropsychologisch onderzoek
Job werd voor het eerst neuropsychologisch onderzocht toen hij 10 jaar was, tenminste 2 jaar na de eerste verschijnselen van de ziekte.
- Gedrag bij onderzoek: stille jongen die goed meewerkt. Aandacht en concentratie zijn duidelijk gestoord. Ten tijde van het onderzoek plast hij in zijn broek.
- Intelligentie: Totaal intelligentiequotiënt (TIQ) = 52, Verbaal IQ = 57, Performaal IQ = 51 (Wechsler Intelligence Scale for Children-Revised).
- Geheugen: Geringe onmiddellijke geheugenspanne voor auditief aangeboden cijfers en voor visueel aangeboden sequenties, passend bij het IQ. In een leertaak (Locaties leren) is het begin zwak en ontstaat hoegenaamd geen leereffect. Bij navertellen van een verhaal confabuleert Job.
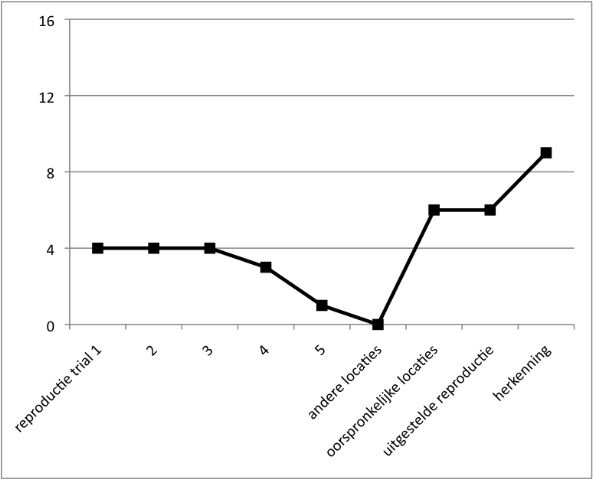
Figuur 3. Locaties leren. - Spraak en taal: De spraak is traag en moeilijk verstaanbaar. Job gebruikt veel stereotype frasen. Hij heeft moeite met vinden van woorden, zo blijkt ook bij benoemen van afbeeldingen. In een zinnendictee maakt Job veel controlefouten. (zie Figuur 4a en b). Bij hardop lezen raakt hij de regel kwijt en slaat hij woorden over.
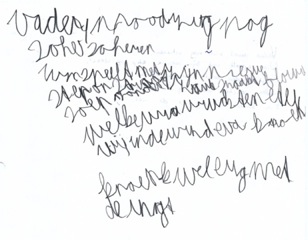
Gedicteerd tijdens onderzoek: vader moet zich nog scheren - soep zonder zout smaakt flauw - welke vruchten eten wij in de winter? - Eva knoeit wel erg met de inkt.

- Rekenen: Bij schriftelijk rekenen lukt optellen slecht en aftrekken erg slecht.
- Grafisch construeren: het vroeger regelmatige handschrift is chaotisch geworden (zie figuur 3); de ooit rijke, fantasievolle tekeningen zijn nu pover.
- Gedragsregulatie: Job kan sorteren maar van sorteerprincipe wisselen lukt niet zonder begeleiding.
- Lichaamschema: intact.
- Reactiesnelheid: Job begrijpt de instructie niet en heeft onvoldoende aandacht voor de taak.
- Handvastheid en handvaardigheid : Bij uitvoeren van taken trillen de handen soms heftig. Coördinatie en differentiatie van het bewegen zijn gestoord.
Laboratoriumonderzoek
Bij enzymonderzoek blijkt arylsulfatase A deficiëntie. De sulfatidefractie in de urine is verhoogd. Bij beeldvorming (MRI) blijken beiderzijds in de witte stof van de hersenen grote confluerende afwijkingen (zie Figuur 5)
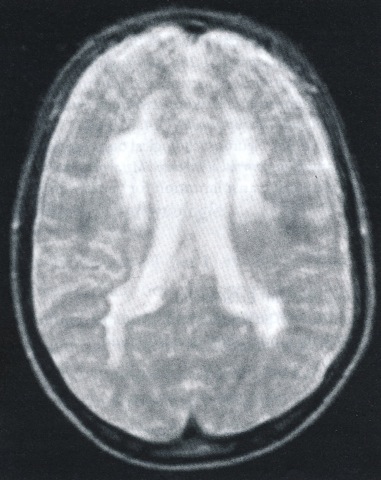
Transversale (dwars, horizontaal)MRI-opname (T2). Het beeld is onscherp door bewegen tijdens het onderzoek. De uitgebreide witte stof afwijkingen beiderzijds in het gebied nabij de hersenkamers zijn duidelijk zichtbaar
Beleid
Omdat de cognitieve stoornissen al ernstig zijn wordt afgezien van stamceltransplantatie. De behandeling is symptomatisch.
Literatuur
LiteratuurBonkowski JL, Nelson C, Kingston JL, Filloux FM, Mundorff MB, Srivastava V (2010) The burden of inherited leukodystrophies. Neurology 75: 718-725
Gieselmann V, Krägeloh-Mann L (2010) Metachromatic leukodystrophy-An update. Neuropediatrics 41: 1-6
Jennekens-Schinkel A, Jennekens FGI (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam: Boom, pp 562-574
Kehrer C, Blumenstock G, Raabe C, Krägeloh-Mann I (2011) Development and reliability of a classification system for gross motor function in children with metachromatic leukodystrophy. Developmental Medicine & Child Neurology 53: 156-160
Pierson M, Bonnemann CG, Finkel RS, Bunin N, Tennekom GI (2008) Umbilical cord blood transplantation for juvenile metachromatic leukodystrophy. Annals of Neurology 64: 583-587
Poorthuis BJHM, Wevers RA, Kleijer WJ, Groener JEM, de Jong JGN, van Weely S, Niezen-Koning KE, Diggelen OP (1999) The frequency of lysosomal storage diseases in the Netherlands. Human Genetics 105: 151-156
Shapiro EG, Lockman LA, Bathazor M, Krivit W (1995) Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. Journal of Inherited Metabolic Diseases 18: 413-429