Myotone dystrofie type 1
Myotone dystrofie type 1 fajenn 25 juni, 2010 - 16:41Inleiding
InleidingMyotone dystrofie type 1 (MD1) is een complexe, zeer langzaam progressieve, erfelijke ziekte. “Myotoon” wijst op vertraagd ontspannen van skeletspieren na aanspanning en ”dystrofie” op onvoldoende volume van skeletspieren. De toevoeging “type 1” geeft aan dat er een aan MD1 verwante ziekte is (MD2). (De Die-Smulders et al. 2010; Jennekens-Schinkel & Jennekens 2008). Hoewel de naam van de ziekte dus past op een spieraandoening, staan bij kinderen jonger dan 10 jaar stoornissen van cognitie en gedrag mede op de voorgrond.
Andere teksten op internet
Andere teksten op internetwww.erfelijkheid.nl. Deze site biedt afzonderlijk informatie over MD1 voor (para)medici en voor patiënten. De site wordt gesteund door de Nederlandse Centra voor Klinische Genetica.
www.vsn.nl. Via de website van de Vereniging Spierziekten Nederland kan men informatie verkrijgen over myotone dystrofie type 1, ook over de aangeboren en kindervormen van de ziekte.
www.kinderneurologie.eu geeft een uitvoerige beschrijving van de ziekte en over de behandeling van ziekteverschijnselen. De tekst is bestemd voor hulpverleners en familieleden.
www.neurologie.nl biedt toegang tot een richtlijn over myotone dystrofie bij volwassenen (18 jaar en ouder).
Epidemiologie
EpidemiologieOnderzoek over de prevalentie in ons land ontbreekt. Mede op grond van gegevens uit andere landen wordt de prevalentie van MD1 in Nederland geschat op 5-15 per 100.000 mensen (de Die-Smulders et al., 2010; www.vsn.nl). Uitgaande van deze cijfers zouden er in Nederland ongeveer 1000-2000 mensen met MD1 zijn. Het aantal kinderen met de ziekte bedraagt waarschijnlijk enige honderden. Of de ziekte bij allochtonen evenveel voorkomt als bij autochtonen is onbekend.
Oorzaak
OorzaakGenmutatie
In een gen op de lange arm van chromosoom 19 ((19q13.3) bevindt zich, in een deel dat voor de coderende functie van het gen niet van belang is, een aantal kopieën van een drietal (een triplet) verschillende nucleotiden die de organische basen cytosine, thymine en guaninine bevatten. Men spreekt daarom van een CTG triplet. Het aantal kopieën (CTG-tripletten) varieert normaal van 5 tot 37 en bij mensen met MD1 van ongeveer 50 tot meer dan 3000 (de Die-Smulders et al., 2010). Hoe groter het aantal kopieën, hoe eerder het debuut van de ziekte en hoe ernstiger de verschijnselen. In opeenvolgende generaties van mensen met de ziekte neemt het aantal kopieën toe.
Het afwijkende gen vertaalt zich in afwijkingen van het RNA. Het gemuteerde RNA hoopt zich op in de kernen en heeft daar een storend effect.
Hersenafwijkingen
Er zijn verschillende - onder de microscoop zichtbare - veranderingen gerapporteerd waarvan niet bekend is of ze algemeen voorkomen en waarvan de betekenis voor de neurologische en neuropsychologische stoornissen onduidelijk is. Met een MRI techniek (DTI, diffusion tensor imaging, zie beeldvorming) zijn consistente afwijkingen in de witte stof zichtbaar gemaakt, zowel bij de kindervormen van de ziekte als bij volwassenen. Een relatie tussen deze witte stof afwijkingen en de cognitieve stoornissen is aannemelijk gemaakt Wozniak et al., 2011; Minnerop et al., 2011)
Erfelijkheid
ErfelijkheidIeder kind van een zieke ouder heeft een kans van 50% op de aandoening. Men noemt deze vorm van overdracht van een ziekte “autosomaal dominante overerving”. Bij MD 1 bestaat anticipatie, (zie figuur stamboom): In opeenvolgende generaties van mensen met MD1 treden de verschijnselen eerder in het leven op. In de eerste generatie zijn de verschijnselen gering en ze manifesteren zich pas op latere leeftijd. In latere generaties zijn de verschijnselen ernstiger en blijken ze vroeger in het leven. Tenslotte zijn de ziekteverschijnselen er vanaf de geboorte. Kinderen met de congenitale, ernstigste vorm van de ziekte worden meestal geboren uit moeders met de ziekte (“maternale” overerving). Een abnormale herhaling (“repeat”) van drie bouwelementen (de nucleotiden cytosine, thymine en guanine) in een gen op chromosoom 19 (19q. 13.3) ligt aan de ziekte ten grondslag. Hoe groter het aantal herhalingen, des te eerder het begin (debuut) van de ziekteverschijnselen. Figuur 1. Stamboom van Familie C. Overgenomen uit de dissertatie van CJ Höweler, 1986.
- I.De man I1 was overleden op het tijdstip dat deze familie werd onderzocht. Hij zou geen verschijnselen van MD1 hebben gehad. Zijn broer I2 zou wel verschijnselen zoals voorkomend bij MD1 hebben gehad.
- II.De vrouw II1 was bij onderzoek in het 7de decennium (tussen 60 en 70 jaar oud); zij had cataract (staar) en verschijnselen van myotonie.
- III.De vrouw III3 had duidelijke verschijnselen van de klassieke vorm van MD1. De vrouw III1 en de man III.2 hadden myotonie sedert de kinderjaren en inmiddels ook spierzwakte; ze waren opvallend passief.
- IV.De vrouw IV1 had de kindervorm van MD1, ze was zwakzinnig en sprak dysarthrisch. De jongen IV2 was overleden, waarschijnlijk ten gevolge van de congenitale vorm van MD1.
- V.Het meisje V1 was overleden ten gevolge van ernstige verschijnselen van de congenitale vorm van MD1.
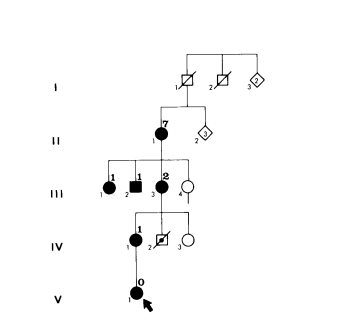
Verklaring: Romeinse cijfers = generaties; grote Arabische cijfers boven gesloten symbolen = levensdecennium waarin MD1 zich openbaarde (leeftijd tussen 60 en 70 jaar); kleine cijfers onder symbolen = persoonsnummer in de studie; vierkant = man; rondje = vrouw; ruit met inwendig cijfer = aantal niet onderzochte gezinsleden; zwarte vulling = MD1 gediagnosticeerd; diagonale streep = overleden; stip in symbool met diagonale streep = dood bij geboorte
Ziekteverschijnselen
ZiekteverschijnselenTabel.De belangrijkste verschijnselen van de vier vormen van MD1
- Debuut vanaf ± 50 jaar – laat debuterende vorm
- staar; soms lichte spierzwakte en myotonie
- Debuut tussen 12 en 45 jaar – zogeheten klassieke vorm
- myotonie, langzaam toenemende (progressieve) spierzwakte, slikstoornissen, trage stoelgang, staar, testikelatrofie (te kleine zaadballen), verhoogde slaapbehoefte, gedragstoornis, ademhalingsstoornissen, hartritmestoornissen,
- Debuut op kinderleeftijd – kindervorm
- intelligentie laag normaal of lichte tot matige zwakzinnigheid, zwakte van gelaat- en keelspieren, onduidelijke spraak, gedragstoornis, vanaf 5de - 10de jaar geleidelijk verschijnselen zoals bij klassieke vorm
- Vanaf de geboorte – congenitale vorm
- spierslapte bij geboorte, zwakte van gelaat- en keelspieren, ernstige zwakzinnigheid tot laag normale intelligentie, gedragstoornis, vanaf 5de tot 10de jaar geleidelijk verschijnselen zoals bij klassieke vorm
Zwakte van de gelaatspieren
Zwakte van gelaat- en keelspieren is vooral bij kinderen met de congenitale vorm maar ook bij kinderen met debuut op kinderleeftijd vroegtijdig aanwezig. Als gevolg daarvan ontwikkelt de aangezichtschedel abnormaal (zie Figuur 2). Het onderste deel van het gelaat is te lang en het gehemelte is hoog en smal. Het gelaat is slap, de mimiek is onvoldoende en de spraak is moeilijk verstaanbaar. Zes en vijftig (56) kinderen en adolescenten (36 met de congenitale vorm, 18 met de kindervorm en twee met de klassieke vorm) hadden allen onvoldoende gelaatsexpressie; bij de meeste kinderen hing de mond in rust open en 28 kinderen hadden dysarthrie (moeilijk verstaanbare spraak) (Sjögreen et al. 2007). Kinderen met MD1 hebben meer plaque, meer tandvleesontsteking en meer cariës dan leeftijdsgenoten (Engvall et al., 2007).

Figuur overgenomen van Jennekens et al., 2000, p 18
[img_assist|nid=922|title=Figuur 2. Zwakte van gelaatspieren bij congenitale vorm van myotone dystrofie type 1.|desc=|link=popup|align=center|width=466|height=640]Figuur overgenomen van Jennekens et al., 2000, p 18
Slaapzucht
Kinderen hebben wat meer slaap nodig dan gebruikelijk, maar “slaapzucht” is, anders dan bij volwassenen met MD1, niet zeer opvallend (Quera Salva et al. 2006).
De diagnose
De diagnose kan definitief worden gesteld door het aantonen van de genetische afwijking. Dit onderzoek wordt verricht als de ziekte- verschijnselen en gegevens over het voorkomen van de ziekte bij een van de ouders daar aanleiding toe geven.
Beloop en medische behandeling
De moeilijke start van kinderen met de congenitale vorm van de ziekte is in variabele mate het gevolg van ademhalingszwakte, slecht zuigen en slikken, onvoldoende motoriek van het maag-darmstelsel en contracturen. Deze verschijnselen kunnen ieder in zekere mate worden opgevangen of tegengegaan door ademhalingsondersteuning, maagsonde voeding, intraveneuze voeding en passief buigen en strekken van gewrichten. De verschijnselen bij de kindervorm van de ziekte kunnen zich gedurende enige tijd beperken tot verminderde mimiek, onvoldoende verstaanbaarheid van de spraak en - vaak - leerstoornissen. Heel geleidelijk ontwikkelen zich zowel bij de congenitale als de kindervorm de verschijnselen van de klassieke vorm. De levensduur wijkt bij de kindervorm en congenitale vorm waarschijnlijk niet veel af van die bij de klassieke vorm, maar hierover zijn nog onvoldoende gegevens. Behandeling van de oorzaak van de ziekte is nog niet mogelijk, wel die van veel ziekteverschijnselen. Denk bijvoorbeeld aan correctie van scheelzien, tandheelkundige zorg, aanpassing van het dieet bij eventuele slikproblemen en stimulering van de stoelgang. Myotone dystrofie type 1 is berucht vanwege de kans op ernstige complicaties bij algemene narcose.
Stoornissen in cognitief functioneren
Stoornissen in cognitief functionerenMatige intelligentie en zwakzinnigheid
Het Totale intelligentiequotiënt (IQ) varieert van lager dan 35 (ernstige zwakzinnigheid) tot – bij uitzondering – boven gemiddeld. Ernstige zwakzinnigheid (grenswaarde van het IQ voor matige en ernstige zwakzinnigheid 50) komt voor bij de congenitale vorm van MD1, vooral bij kinderen met een na de geboorte erg moeilijke start (ENCYCL-Zwakzinnigheid). Slechts weinig kinderen met de congenitale vorm hebben een laag normale intelligentie (IQ varieërt van meer dan 70 tot 85) en heel weinige een normale intelligentie (Ekström et al. 2008; 2009). Bij de kindervorm van MD1 is het Totale IQ gemiddeld lager dan in de gehele bevolking. Een gemiddelde heeft voor het individuele kind weinig betekenis wanneer de spreiding zo groot is als bij deze vorm van MD1 (zie figuur, ontleend aan Angeard et al. 2007). Zo was in een groep van 34 kinderen het Totale IQ gemiddeld 69,8. Maar het laagste IQ was 42, passend bij matige zwakzinnigheid. En het hoogste 114, passend bij een boven gemiddelde intelligentie (Angeard et al. 2007). Eerder was bij 24 kinderen een niet wezenlijk ander beeld gevonden (Steyaert et al. 2000).
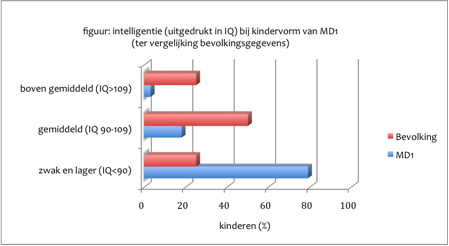
[img_assist|nid=1208|title=Staafdiagram op basis van gegevens van Angeard et al., 2007 |desc=|link=popup|align=center|width=480|height=263] Het Verbale IQ is bij de kindervorm misschien iets hoger dan het Performale IQ maar het verschil is niet groot en het aantal kinderen waarvoor deze gegevens beschikbaar zijn is beperkt zodat een definitieve uitspraak nog niet mogelijk is. Bij overerving via de moeder is het Totale IQ gemiddeld wat lager dan bij paternale (via de vader) overerving.
Stoornissen in andere cognitieve functies
Specifieke tekorten in andere domeinen van het cognitief functioneren zijn niet gevonden of nog onvoldoende bekend om conclusies te kunnen trekken. Geheugen en leervermogen zijn naar verhouding sterk. Traagheid van denken is geopperd maar niet bevestigd (zie Jennekens-Schinkel en Jennekens, 2008).
Stoornissen in gedragfuncties
Stoornissen in gedragfunctiesAutisme of op autisme lijkende verschijnselen
Tot de kernverschijnselen van autisme behoren onvoldoende sociale interactie, onvoldoende communicatie en stereotyp (telkens terugkerend) gedrag (ENCYCL-Autisme). Voor het vaststellen van autisme bestaat geen laboratoriumproef die de resultaten van vragenlijstonderzoek, interview en algemene indruk kan toetsen (Spence & Thurm, 2010). Dat vergroot de kans op een subjectief element bij het vaststellen van de stoornis. Kinderen met congenitale MD1 communiceren moeilijk door de gelaatspierzwakte en de spraakstoornis (dysartrie). In één studie werd tot autisme geconcludeerd bij iets meer dan één op de twee kinderen met de congenitale vorm van MD1 en bij één op de zes kinderen met de kindervorm, vooral op grond van onvoldoende sociale interactie en onvoldoende communicatie (Ekström et al. 2008). Het zou kunnen dat de criteria van autisme niet scherp zijn onderscheiden van de verschijnselen van MD1.
Stoornis in temperament
Volwassenen met MD1 hebben volgens hun omgeving weinig initiatief, zij zijn weinig fel in hun reageren op prikkels en zij hebben een laag energie- en activiteitsniveau. Zij hebben, kortom, een nog niet begrepen afwijking van het temperament (het hoe van gedrag, de gedragsstijl). Op een temperamentlijst scoorden zij extreem laag wanneer het ging om behoefte aan nieuwe ervaringen en avontuur. Andere dimensies van het temperament (extraversie, emotionaliteit, impulsiviteit) waren ongestoord. (Jennekens-Schinkel & Jennekens, 2008).
Het gedrag van kinderen wordt beschreven als kalm, neigend tot passiviteit, ze nemen weinig initiatieven voor het komen tot contact, ze zijn niet snel afgeleid, ze passen zich goed aan en ze zijn vriendelijk (Ekström et al., 2010; Steyaert et al., 1997)
Psychosociaal functioneren
Psychosociaal functionerenSchoolloopbaan
Omstreeks 50% of minder van de kinderen met de kindervorm van MD1 kan met extra onderwijskundige hulp regulier onderwijs volgen. De overigen behoeven speciaal onderwijs evenals kinderen met de congenitale vorm (Quera Salva et al., 2006).
Sociale interactie
De slapte van het gezicht doet afbreuk aan de mogelijkheden tot het uiten van emoties. De onduidelijkheid van de spraak belemmert de communicatie. In sport scoren de kinderen slecht. Hun passiviteit en - bij sommigen - de verhoogde slaapbehoefte worden niet altijd begrepen als ziekteverschijnselen. Autisme kan het tekort in interactie nog versterken. Meer dan andere kinderen staan zij bloot aan pesterijen.
Begeleiding
BegeleidingDe combinatie van lichamelijke-, cognitieve- en gedragstoornissen, en het uiterst langzame progressieve beloop stellen hoge eisen aan de begeleiding.
- Ouders kunnen zich bezwaard voelen door het besef van hun verantwoordelijkheid voor de ziekte van hun kind en kunnen om die reden bijstand nodig hebben. Wat ook de beperkingen van de kinderen zijn, ze moeten zoveel als mogelijk tot zelfstandige mensen worden opgevoed
- Ouders, leraren en andere begeleiders behoeven informatie over de vele ongewone aspecten van de ziekte: de gevolgen van de spierzwakte in het gelaat voor mimiek en verstaanbaarheid van de spraak, de kans op zeer verschillende lichamelijke klachten, de passieve instelling van de kinderen, de meerdere slaapbehoefte van sommige kinderen, de beperkingen die de ziekte geeft in sport en spel, het risico dat de kinderen worden gepest en het risico dat de kinderen ten onrechte als lui worden beschouwd.
- Onderzoek van de cognitie kan gewenst zijn voor een verantwoord advies over onderwijs en schoolkeuze. De gelaatsuitdrukking van kinderen kan een onjuiste schatting van de cognitieve mogelijkheden in de hand werken.
- Begeleiders moeten zo nodig ouders stimuleren het kind tenminste eenmaal per jaar medisch te laten evalueren.
Literatuur
LiteratuurAngeard N, Gargiulo M, Jacquette A, Radvanyi H, Eymard B, Héron D. (2007) Cognitive profile in childhood myotonic dystrophy type 1: Is there a global impairment? Neuromuscular Disorders 17: 451-458
De Die-Smulders CEM, Jennekens FGI, Faber CG (2010) Myotonic dystrophy type 1. In: Cassidy SB, Allanson JE (redacteuren) Management of genetic syndromes. 3de druk, Wiley & Sons Inc. pp 529-547
Ekström AB, Hakenäs-Plate L, Samuelsson L, Tulinius M, Wentz E. (2008) Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. American Journal of Medical Genetics B Neuropsychiatric Genetics 147B : 918-926
Ekström AB, Hakenās-Plate L, Tulinius M, Wentz E (2009) Cognition and adaptive skills in myotonic dystrophy type 1: a study of 55 individuals with congenital and childhood forms. Developmental Medicine & Child Neurology 51: 982-990
Engvall M, Sjögreen L, Kjellberg H, Robertson A, Sundall S, Killarides S. (2007). Oral health in children and adolescents with myotonic dystrophy. European Journal of Oral Science 15; 192-197
Jennekens FGI, De Die-Smulders CEM, Busch HFM, Höweler CJ. (2000) Myotone dystrofie begeleiding en behandeling. Maarssen: Elsevier Gezondheidszorg
Jennekens-Schinkel A, Jennekens FGI. (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam: Uitgeverij Boom, pp 575-590
Minnerop M en 10 andere auteurs. (2011) The brain in myotonic dystrophy type 1 and 2: evidence for a predominant white matter disease. Brain 134: 3530-3546
Quera Salva M-A, Blumen M, Jacquette A, Durand M-C, Andre S, Villiers M de et al. (2006) Sleep disorders in childhood-onset myotonic dystrophy type 1. Neuromuscular Disorders 2006; 16: 564-570
Sjögreen L, Engvall M, Ekström A, Lohmander A, Kiliaridis S, Tulinius M. (2007) Orofacial dysfunction in children and adolescence with myotonic dystrophy. Developmental Medicine and Child Neurology 2007; 49: 18-22
Spence SJ, Thurm A. (2010) Testing autism interventions:trials and tribulations. Lancet 375: 2124-2125
Steyaert J, De Die-Smulders C, Frijns J-P, Goossens E, Willekens D. (2000) Behavioral phenotype in childhood type of dystrophia myotonica. American Journal of Medical Genetics (Neuropsychiatric Genetics) 96: 888-889
Wozniak JR, Mueller BA, Ward EE, Lim KO, Day JW. (2011) White matter abnormalities and neurocognitive correlates in children and adolescents with myotonic dystrophy type 1: a diffusion tensor imaging study. Neuromuscular Disorders 21: 89-96